
 Benchmarking spatial clustering methods with spatially resolved transcriptomics data
Benchmarking spatial clustering methods with spatially resolved transcriptomics data
题目: 使用空间解析的转录组数据对空间聚类方法进行基准测试
DOI: https://doi.org/10.1038/s41592-024-02215-8 (opens new window)
Cite: Yuan, Z., Zhao, F., Lin, S. et al. Benchmarking spatial clustering methods with spatially resolved transcriptomics data. Nat Methods 21, 712–722 (2024).
作者介绍:
| Yi Zhao |
|---|
 |
| 中国科学院大学 |
| qbiozy@ict.ac.cn |
# Abstract:
空间聚类与单细胞聚类类似,它将组织生理学研究的范围从细胞中心扩展到结构中心,并利用空间转录组(SRT)数据。近年来,计算方法取得了显著发展,但仍缺乏全面的基准研究。本文对13种计算方法在34个SRT数据(7个数据集)上进行了基准测试。评估指标包括准确性、空间连续性、标记基因检测、可扩展性和鲁棒性。我们发现现有方法在性能和功能上具有互补性,并为特定场景下的方法选择提供了指导。在测试另外22个具有挑战性的数据集时,我们发现了识别非连续空间域的挑战以及现有方法的局限性,突出了它们在处理近期大规模任务时的不足。此外,利用145个模拟数据,我们检验了这些方法对四种不同因素的鲁棒性,并评估了预处理和后处理方法的影响。我们的研究对现有空间聚类方法在SRT数据上的表现进行了全面评估,为这一快速发展领域的未来进步铺平了道路。
# Results:
# Figure 1: Pipeline and data.
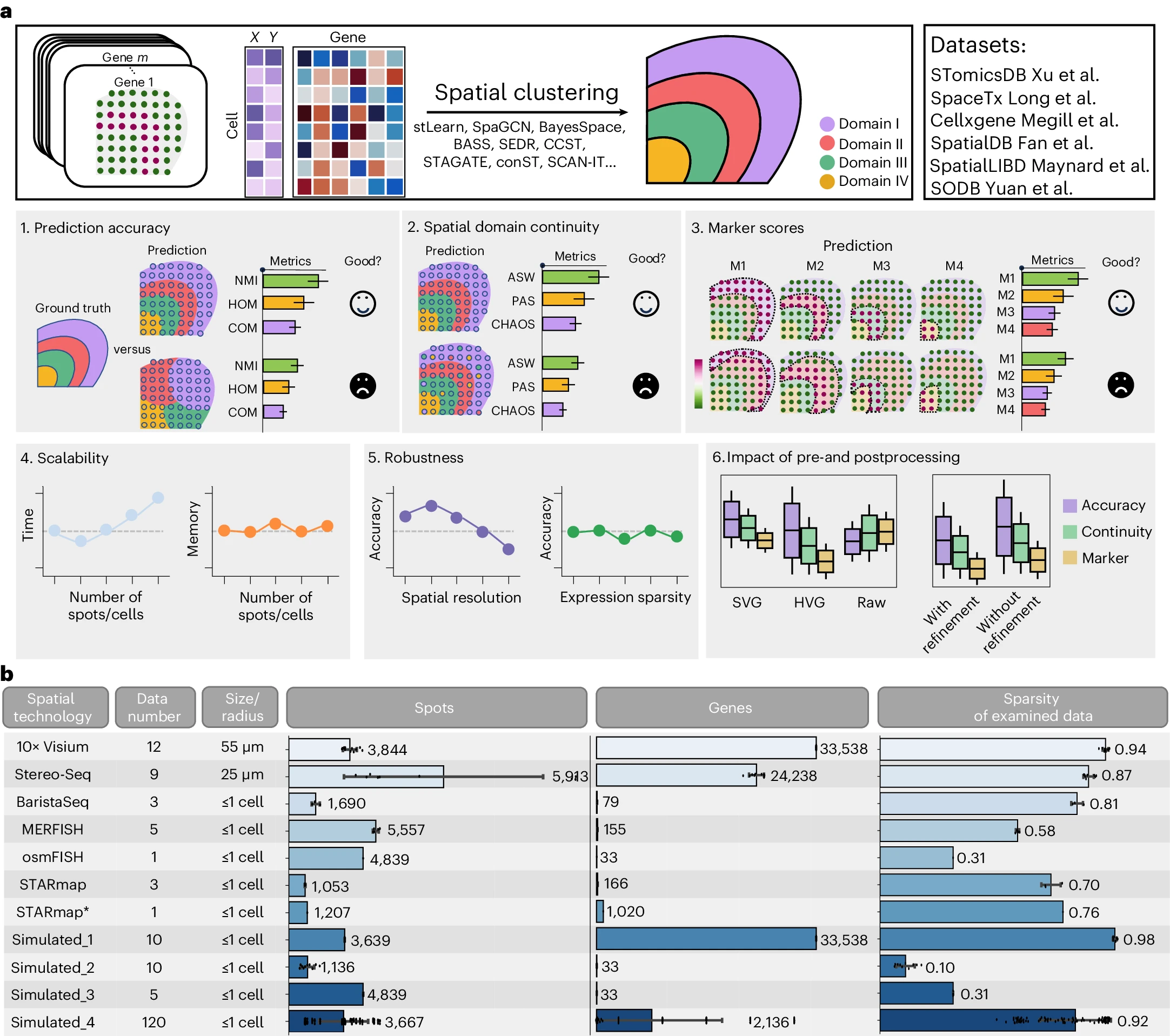
Figure 1. 流程和数据
(a) Benchmark研究的流程。
(b) 本研究中使用的数据集。列出了子数据集的数量、空间分辨率、spot/细胞数量、基因数量和基因表达矩阵稀疏度等信息。请注意,条形的长度与平均值成比例,条形旁边的数字是中值。
# Figure 2: Evaluation on 10x Visium and MERFISH.
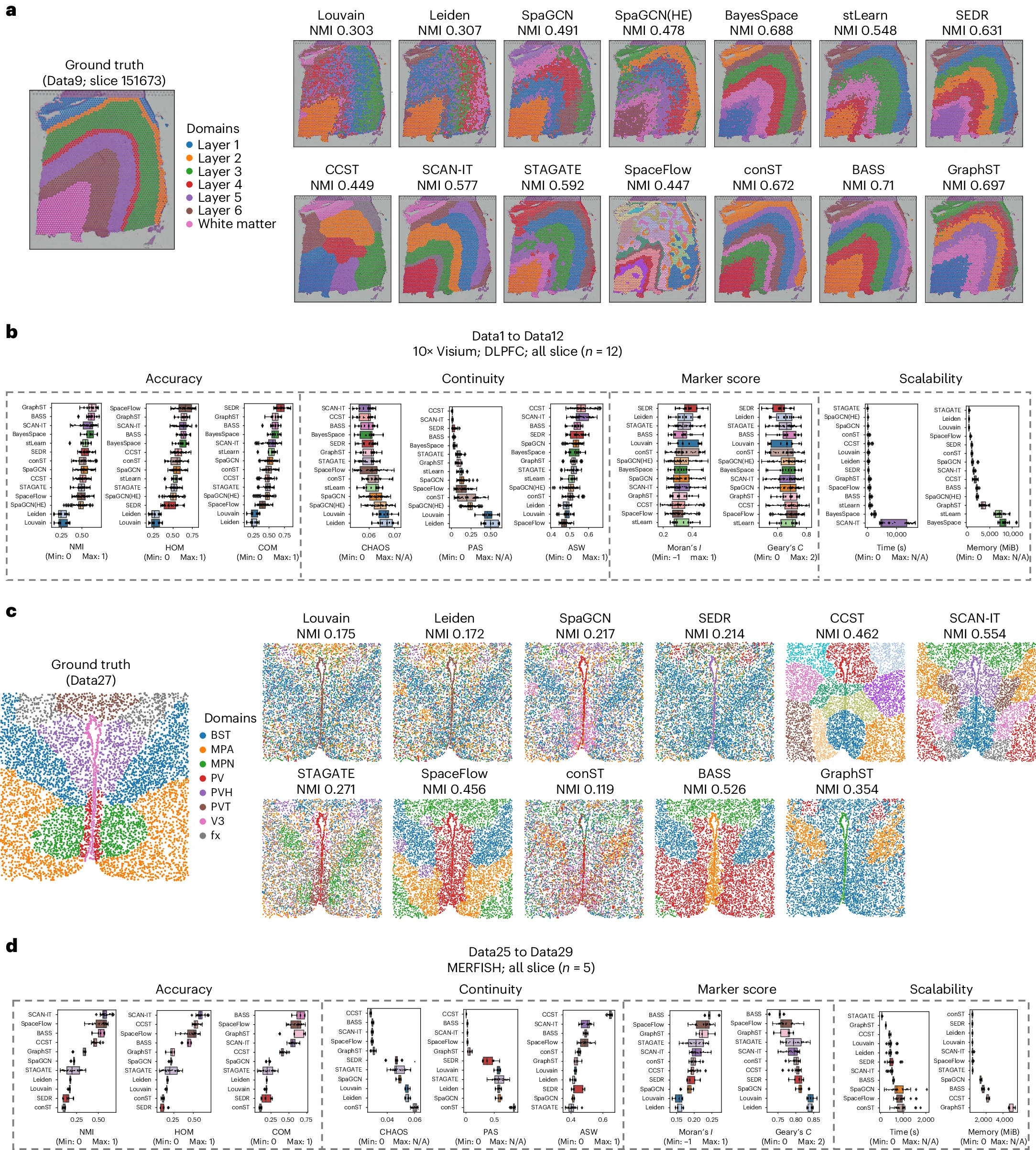
Figure 2. 对 10x Visium 和 MERFISH 的评估
(a) 10x Visium 数据集的代表性切片上,展示了“真实”标准与各方法的输出结果。
(a) 在所有 10x Visium 数据的 12 组数据上进行了定量评估。对于每组数据,每种方法均重复运行 10 次。
(c) MERFISH 数据集的代表性切片上,展示了“真实”标准与各方法的输出结果。
(d) 数据集 4 中转录本的 8 种整合方法的 AS 小提琴图(从 PCC、SSIM、RMSE 和 JS 值聚合;参见方法);n = 4 个基准指标。
(e) 在所有 MERFISH 数据的 5 组数据上进行了定量评估。对于每组数据,每种方法均重复运行 10 次。
# Figure 3: Correlations between data regarding methods performance.
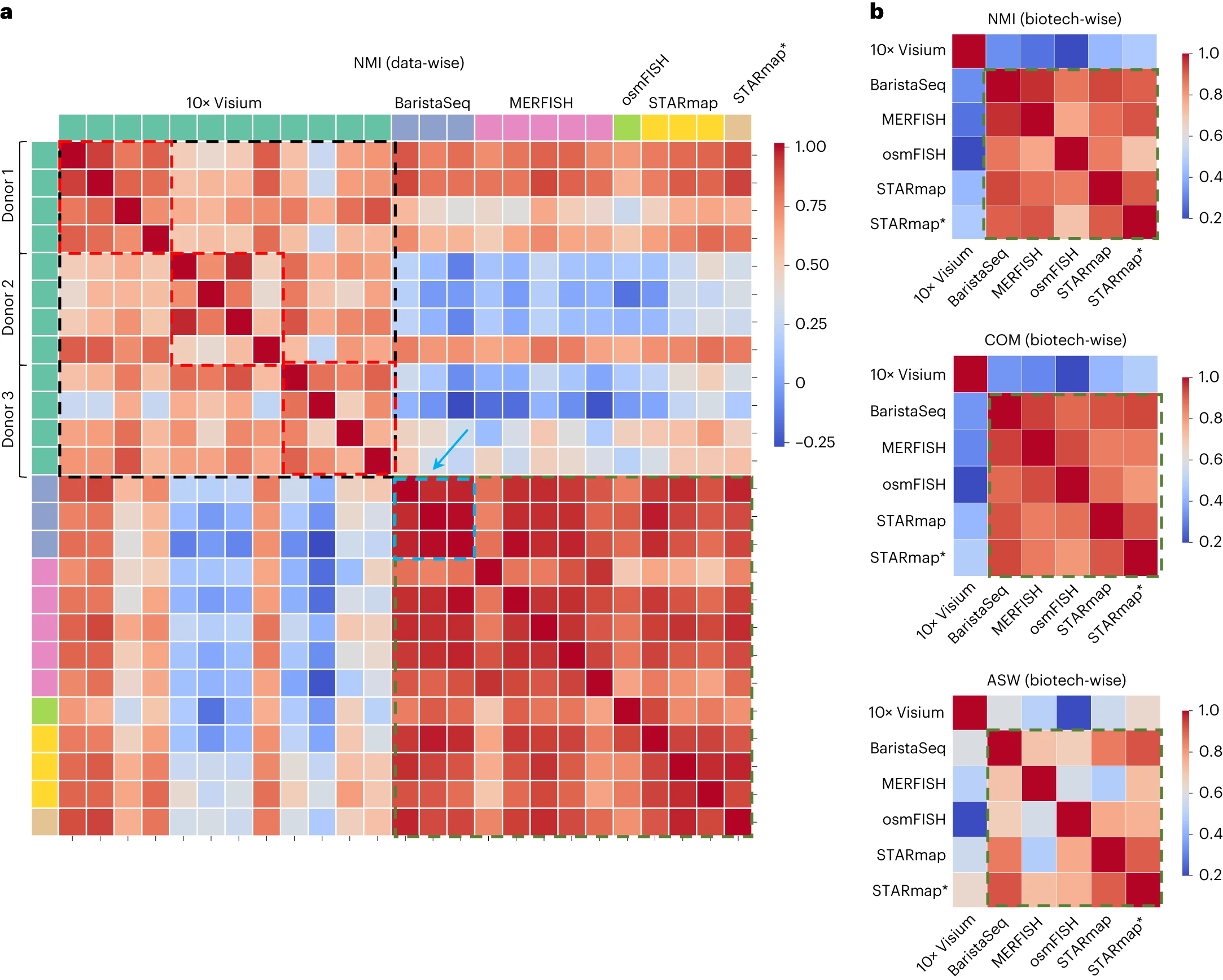
Figure 3. 方法性能数据之间的相关性
(a) 在热图中,行 i 和列 j 的条目是通过计算两个向量的 Spearman 相关性得到的。其中一个向量由所有方法在数据 i 上的中位 NMI 组成,另一个向量则由所有方法在数据 j 上的中位 NMI 组成。黑色虚线框表示 10x Visium 数据之间的相关性。绿色虚线框表示基于成像的 SRT 数据之间的相关性。蓝色箭头指向的蓝色虚线框表示 BaristaSeq 数据之间的相关性。在 10x Visium 数据集中,数据来自三位供体,红色虚线框表示来自同一供体的数据之间的相关性。
(b) 与 a 类似,但相关性分析的对象是生物技术之间,而非数据。绿色虚线框表示基于成像的 SRT 技术之间的相关性。
# Figure 4: Overall performance.
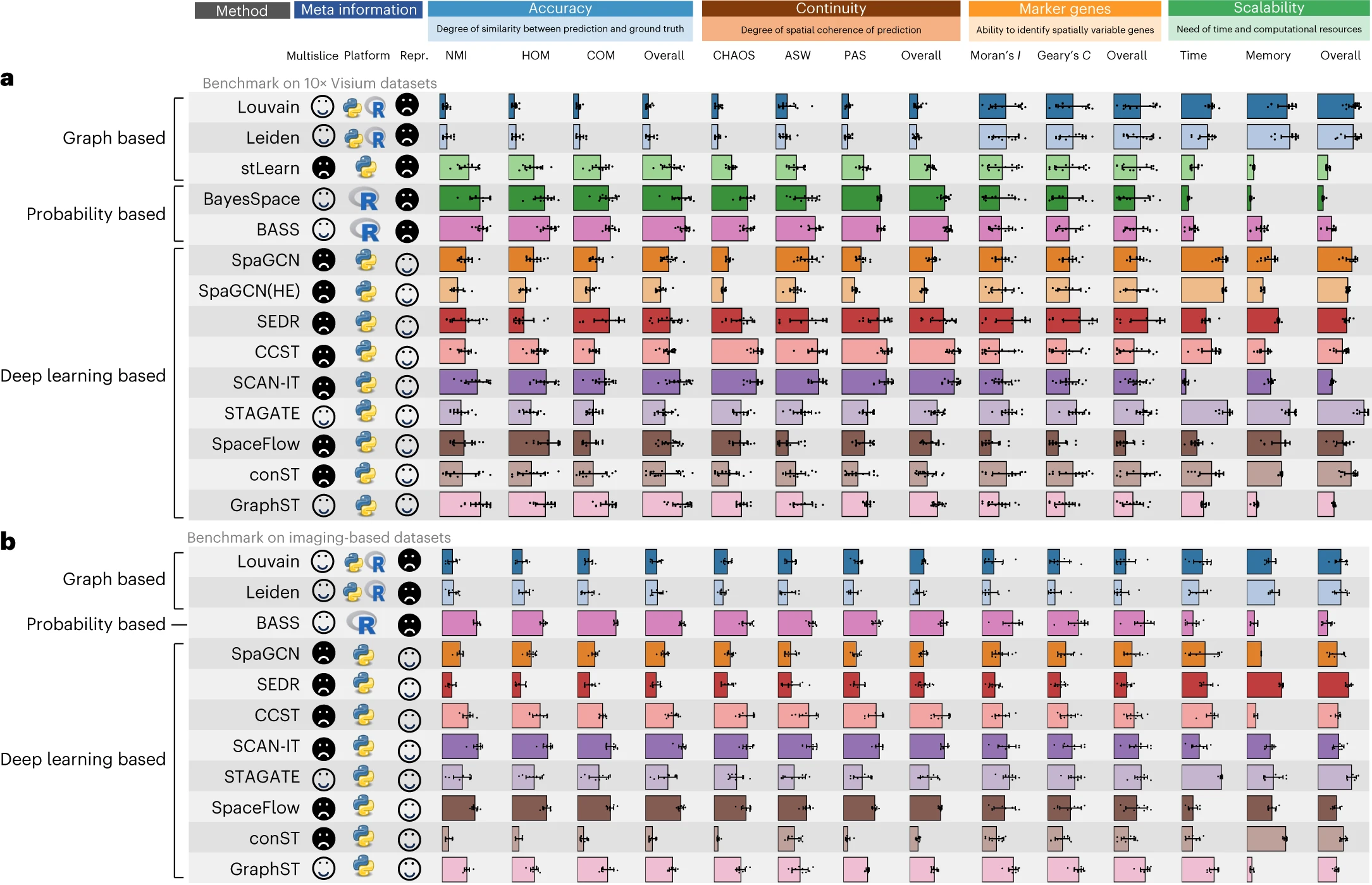
Figure 4. 整体性能
(a,b) 方法信息包括多切片分析支持(是否支持多切片分析)、平台(Python 或 R)、repr.(是否能够输出上下文感知表示)、NMI、HOM、COM、CHAOS、ASW、PAS、Moran’s I、Geary’s C、运行时间和峰值内存。这些指标采用基于排名的分数进行展示(见方法部分)。所应用的数据集分为两组,即 10x Visium(a)和基于成像的 SRT(b)。
# Figure 5: The large-scale datasets are solved by proposed approach.
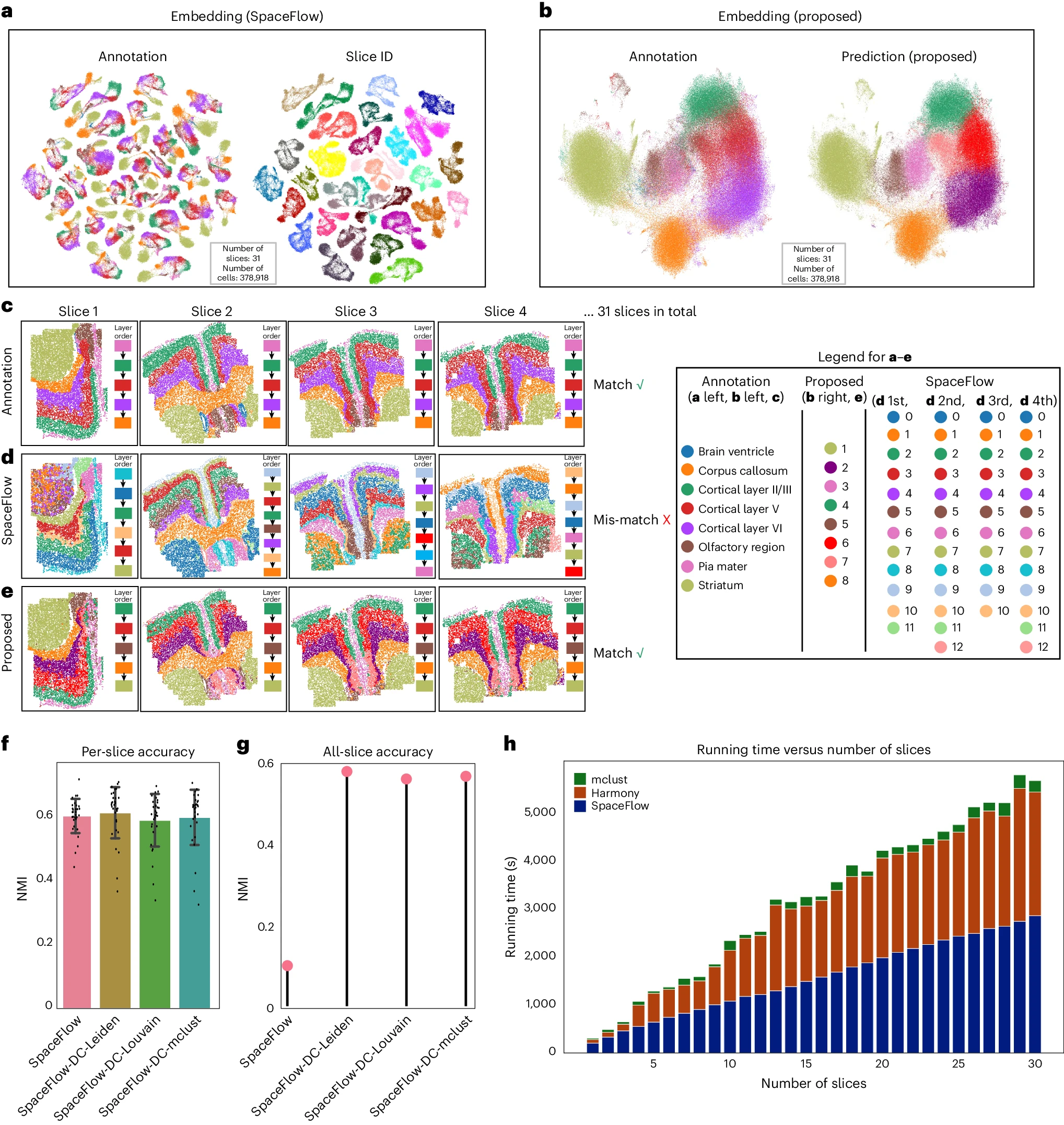
Figure 5. 通过提出的方法解决大规模数据集。
(a) 该可视化图通过对所有切片的 SpaceFlow 嵌入应用 UMAP 生成,左图按真实标注(ground truth)着色,右图按切片 ID 着色。
(b) 该可视化图通过对所有切片的 SpaceFlow-DC 嵌入(本文提出的方法)应用 UMAP 生成,左图按真实标注着色,右图按SpaceFlow-DC 预测的域标签着色。
(c) 专家标注的真实标签在不同切片间保持一致。
(d) 原始 SpaceFlow 方法无法在不同切片间生成对齐的标签。
(e) SpaceFlow-DC 生成的域标签在不同切片间匹配良好。
(f) 记录了每个切片上,SpaceFlow 和三种版本的 SpaceFlow-DC(本文提出的方法)的 NMI(归一化互信息)值,并以均值和 95% 置信区间展示(N = 31 个切片)。
(g) 记录了所有切片上的 NMI 值,对比 SpaceFlow 和三种版本的 SpaceFlow-DC。
(h) 记录了 SpaceFlow-DC 在不同切片子集上三个不同步骤(SpaceFlow、Harmony 和 mclust)的运行时间。
# Figure 6: The impact of pre- and postprocessing.
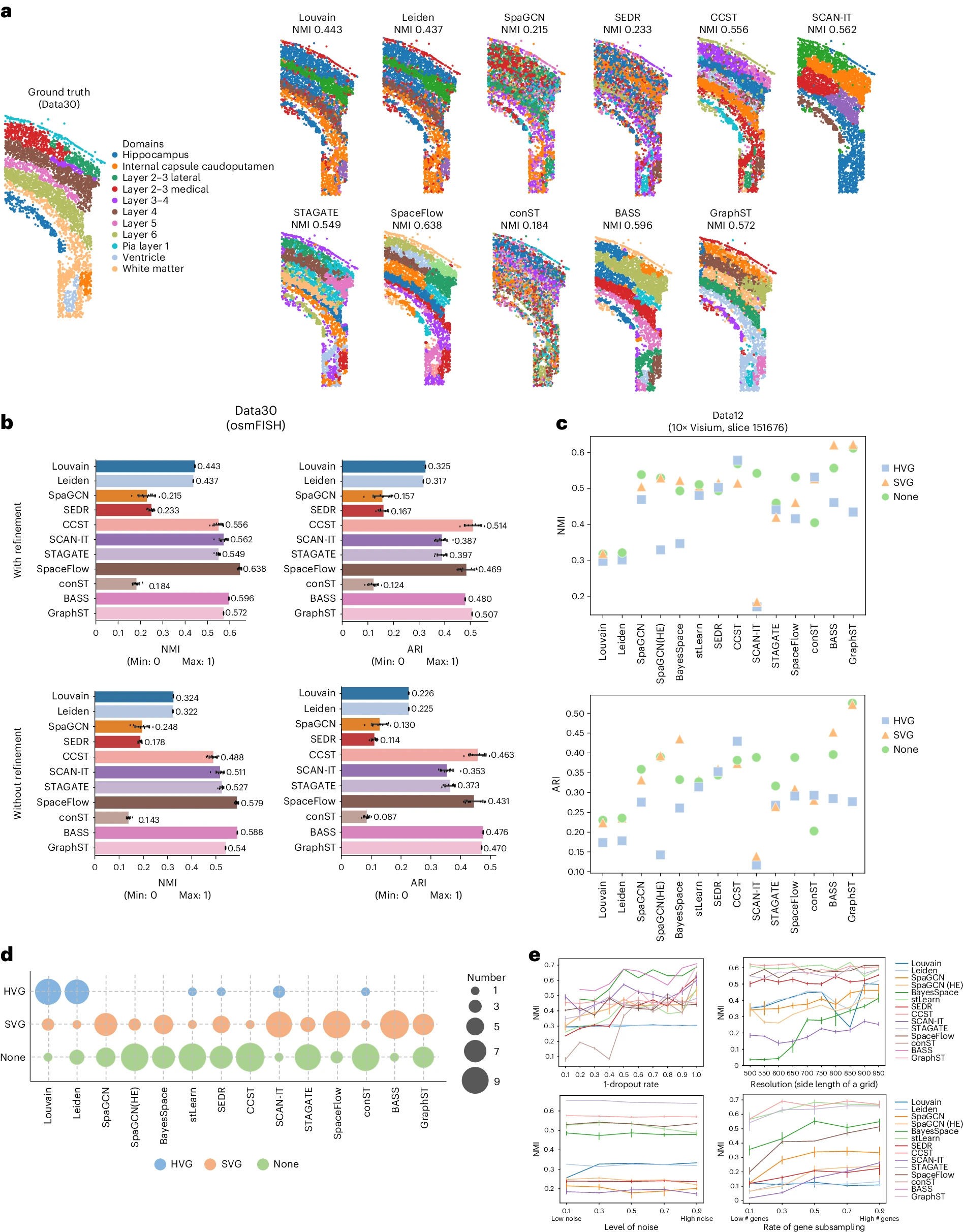
Figure 6. 预处理和后处理的影响。
(a) osmFISH 数据上的真实标签(ground truth)与不同方法的输出结果。所有结果均经过基于 KNN 的优化方法进行后处理。。
(b) 在 osmFISH 数据上记录了不同方法的定量评估,分别展示了经过优化(上图)和未经过优化(下图)的结果。数据以均值和 95% 置信区间表示(N = 10 次独立运行)。
(c) 在 10x Visium 数据上记录了不同预处理方法的影响,包括高变基因(HVG)筛选、空间可变基因(SVG)筛选以及无预处理(None)。
(d) 不同预处理方法下各方法的表现。气泡图展示了不同预处理方法与空间聚类方法的组合表现。点的大小表示在多少个数据集中,该预处理方法的 NMI 值高于其他两种预处理方法。
(e) 方法在四个不同因素下的稳健性评估:左上:基因表达矩阵的稀疏程度, 右上:空间分辨率, 左下:噪声水平, 右下:基因数量
# Discussion:
本研究对13种空间转录组数据聚类方法进行了全面的基准分析,评估了其在准确性、连续性、标记基因评分和可扩展性方面的表现。研究发现,没有一种方法在所有数据集上表现最佳,最佳方法的选择高度依赖于数据特征。当前方法在小区域识别、多切片分析和大规模数据处理方面存在显著局限性。为解决这些问题,研究提出了一种“分而治之”策略,通过结合现有工具有效应对大规模数据集的挑战。然而,由于空间转录组领域的快速发展和真实标注数据的有限性,某些新兴数据类型未被纳入研究,且研究主要集中于空间转录组数据,未涵盖其他空间数据类型(如空间蛋白质组学和空间多组学数据)。未来研究需要进一步扩展数据类型的覆盖范围,并优化方法以提升鲁棒性和可扩展性。