
 Integration of spatial and single-cell data across modalities with weakly linked features
Integration of spatial and single-cell data across modalities with weakly linked features
题目: 具有弱关联特征的跨模态空间和单细胞数据的整合
DOI: https://doi.org/10.1038/s41587-023-01935-0 (opens new window)
Cite:Chen, S., Zhu, B., Huang, S. et al. Integration of spatial and single-cell data across modalities with weakly linked features. Nat Biotechnol (2023).
作者介绍:
| Zongming Ma |
|---|
 |
| Department of Statistics and Data Science, Yale University, New Haven, CT, USA |
| zongming.ma@yale.edu |
# Abstract:
尽管单细胞和空间测序方法能够同时测量多种生物模态,但没有任何技术可以捕获同一细胞内的所有形态。对于当前的数据集成方法,跨模式集成的可行性依赖于高度相关、先验“链接”特征的存在。我们描述了通过模糊平滑嵌入(MaxFuse)匹配X模态,这是一种跨模态数据集成方法,通过迭代共嵌入、数据平滑和单元匹配,即使在特征很弱的情况下,也可以使用每种模态中的所有信息来获得高质量的集成已链接。MaxFuse 与模态无关,在弱链接场景中表现出高鲁棒性和准确性,在基准数据集的关键评估指标下比现有方法实现了 20~70% 的相对改进。弱关联的一个典型例子是空间蛋白质组数据与单细胞测序数据的整合。在此类类型的两个示例分析中,MaxFuse 能够在同一组织切片上以单细胞分辨率对蛋白质组、转录组和表观基因组信息进行空间整合。
# Results:
# Figure 1: Overview of MaxFuse pipeline.
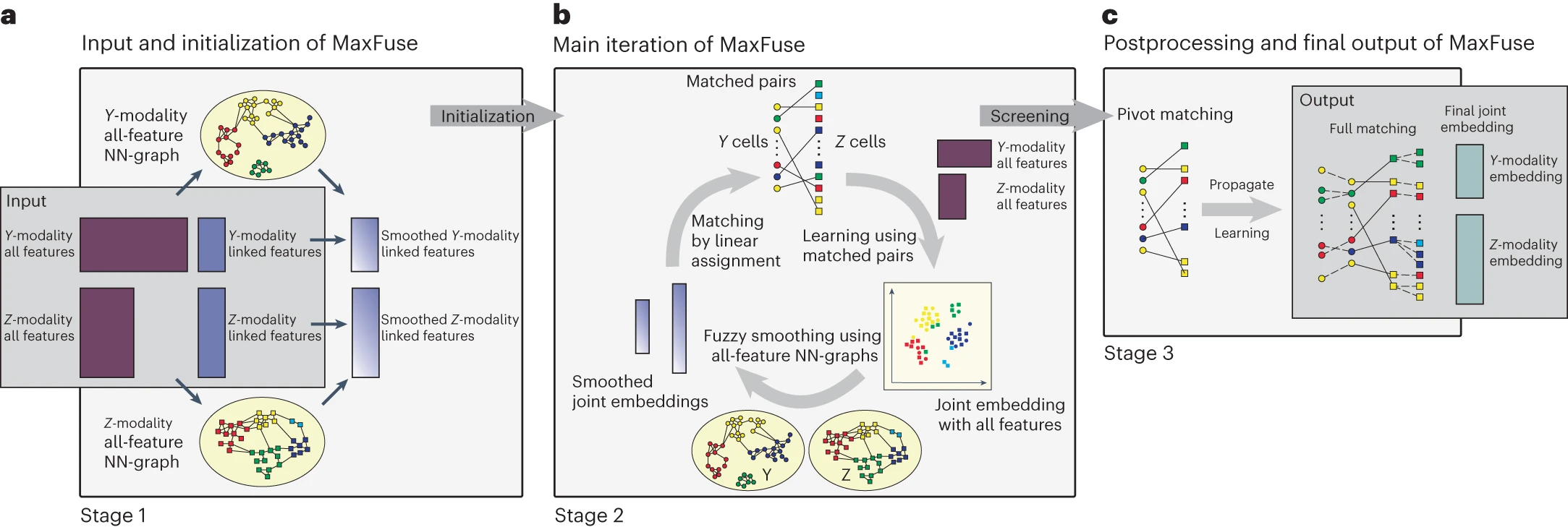
Figure 1. MaxFuse 流程概述
(a) 输入由两对矩阵组成。第一对由每种模态的所有特征组成,第二对仅由链接的特征组成。MaxFuse 使用每种模态中的所有特征为该模态中的单元创建最近邻图(即全特征神经网络图)。由全特征神经网络图引起的模糊平滑被应用于每种模态中的链接特征。基于平滑链接特征的跨模式单元匹配初始化b中的迭代。
(b) 在每次迭代中,MaxFuse 从匹配单元对的列表开始。跨模式单元对称为枢轴。MaxFuse 基于这些枢轴学习两种模式的所有特征的典型相关分析 (CCA) 负载。这些 CCA 负载允许计算每个单元(包括不在任何枢轴中的单元)的 CCA 分数,这些分数用于获得跨两种模式的所有单元的联合嵌入。对于每种模态,嵌入坐标随后根据模态特定的全特征神经网络图(在a中获得)进行模糊平滑。接下来,将平滑的嵌入坐标提供给线性分配算法,该算法生成更新的匹配对列表以开始下一次迭代。
(c) 迭代结束后,MaxFuse 筛选最终的枢轴列表以删除低质量匹配。保留的对称为精化主元。在每种模态中,不属于精炼枢轴一部分的任何单元都连接到属于精炼枢轴的其最近邻居,并与该枢轴中其他模态的单元相匹配。此传播步骤导致完全匹配。MaxFuse 基于精炼的枢轴进一步了解两种模式所有功能的最终 CCA 负载。由此产生的 CCA 分数给出了最终的联合嵌入坐标。
# Figure 2: Benchmarking of MaxFuse and other integration methods on ground-truth CITE-seq PBMC data.
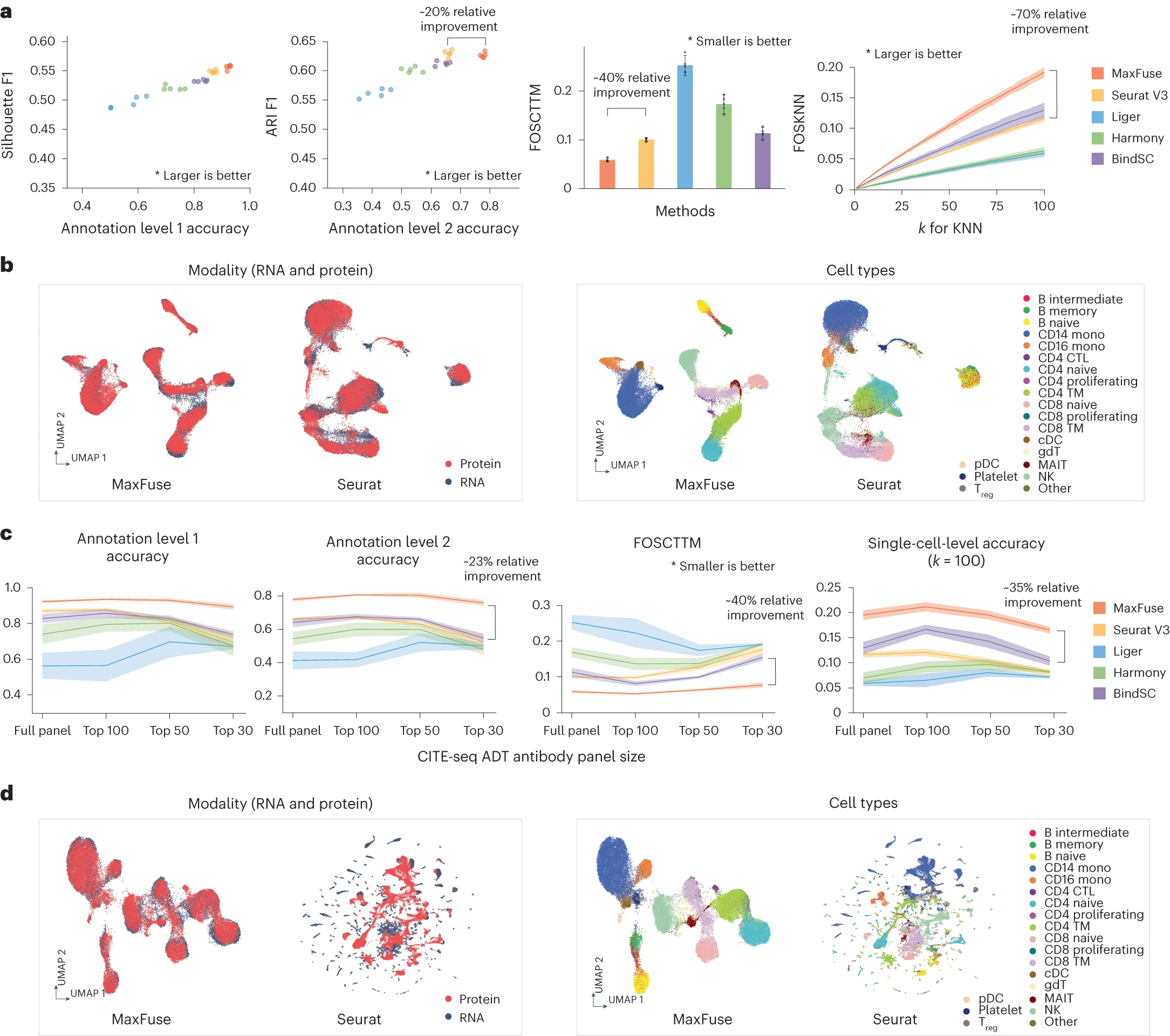
Figure 2. MaxFuse 和其他集成方法在真实 CITE-seq PBMC 数据上的基准测试
(a) MaxFuse 和其他方法在 CITE-seq PBMC 数据集上与完整抗体组(228 种抗体)的匹配和集成性能。条形图和线图显示平均值,误差条或阴影区域两侧均覆盖 95% CI,来自n = 5 个随机二次采样的细胞批次。
(b) CITE-seq PBMC 数据集的 MaxFuse 和 Seurat (V3) 集成结果的 UMAP 可视化与完整面板,按模态(左)或细胞类型(右)着色。
(c) MaxFuse 和其他方法在具有精简抗体组(完整 228 个抗体或信息最丰富的 100、50 或 30 个抗体)的 CITE-seq PBMC 数据集上的匹配和集成性能。
(d) CITE-seq PBMC 数据集的 MaxFuse 和 Seurat (V3) 整合结果的 UMAP 可视化,其中包含原始 228 种抗体中信息最丰富的 30 种,按形态(左)或细胞类型(右)着色。
# Figure 3: Benchmarking of MaxFuse versus other integration methods across multiple ground-truth data types.
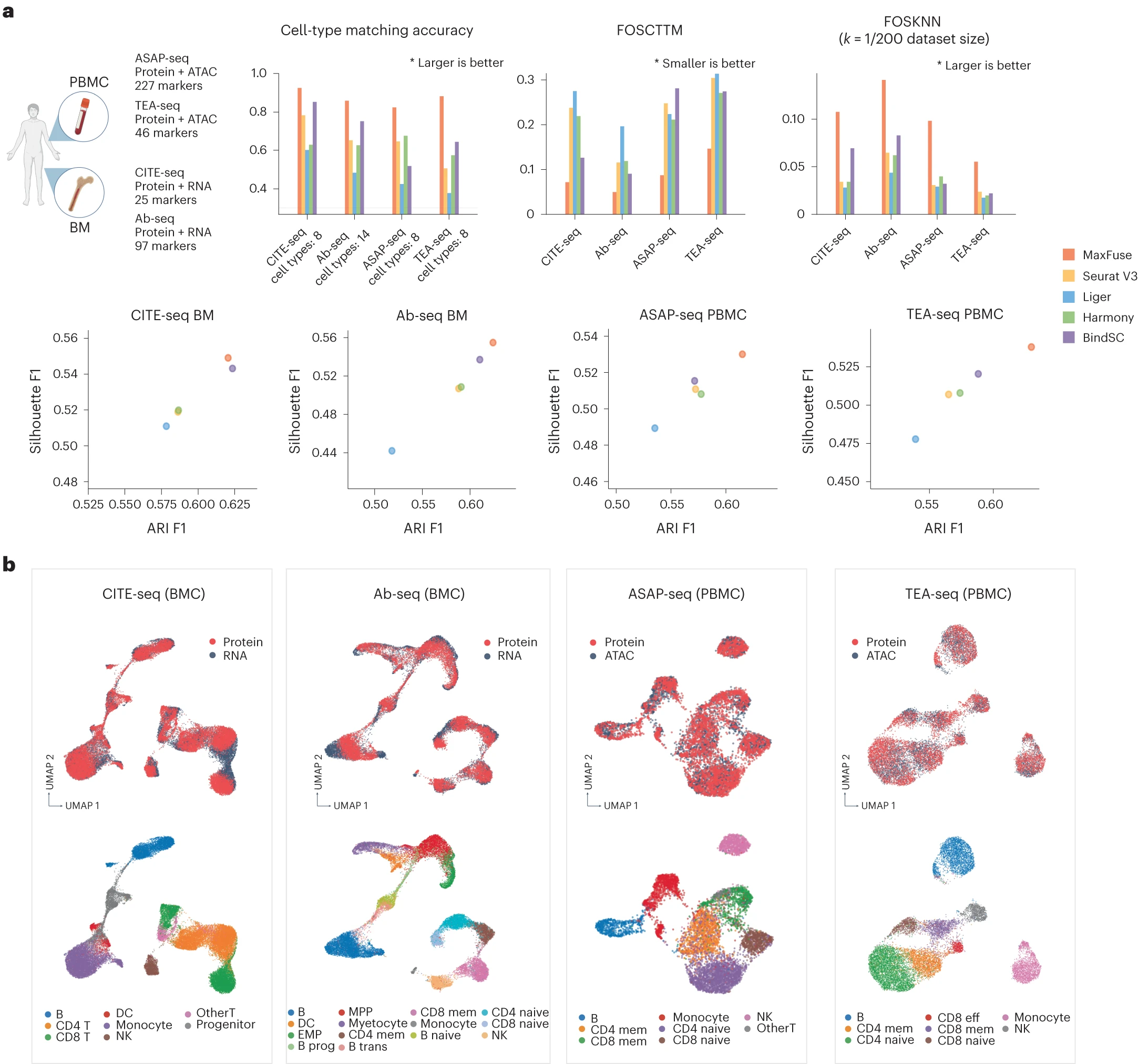
Figure 3. MaxFuse 与跨多种真实数据类型的其他集成方法进行基准测试
(a) 对由不同技术生成的四个不同的多组数据集进行了基准测试。细胞类型匹配准确性、FOSCTTM、FOSKNN 以及 ARI 和 Silhouette F1 在所有五种方法中进行了评估。
(b) 四个真实多组数据集的 MaxFuse 集成结果的 UMAP 可视化,按模态(顶部面板)和细胞类型(底部面板)着色。
# Figure 4: MaxFuse enables information-rich spatial pattern discovery.
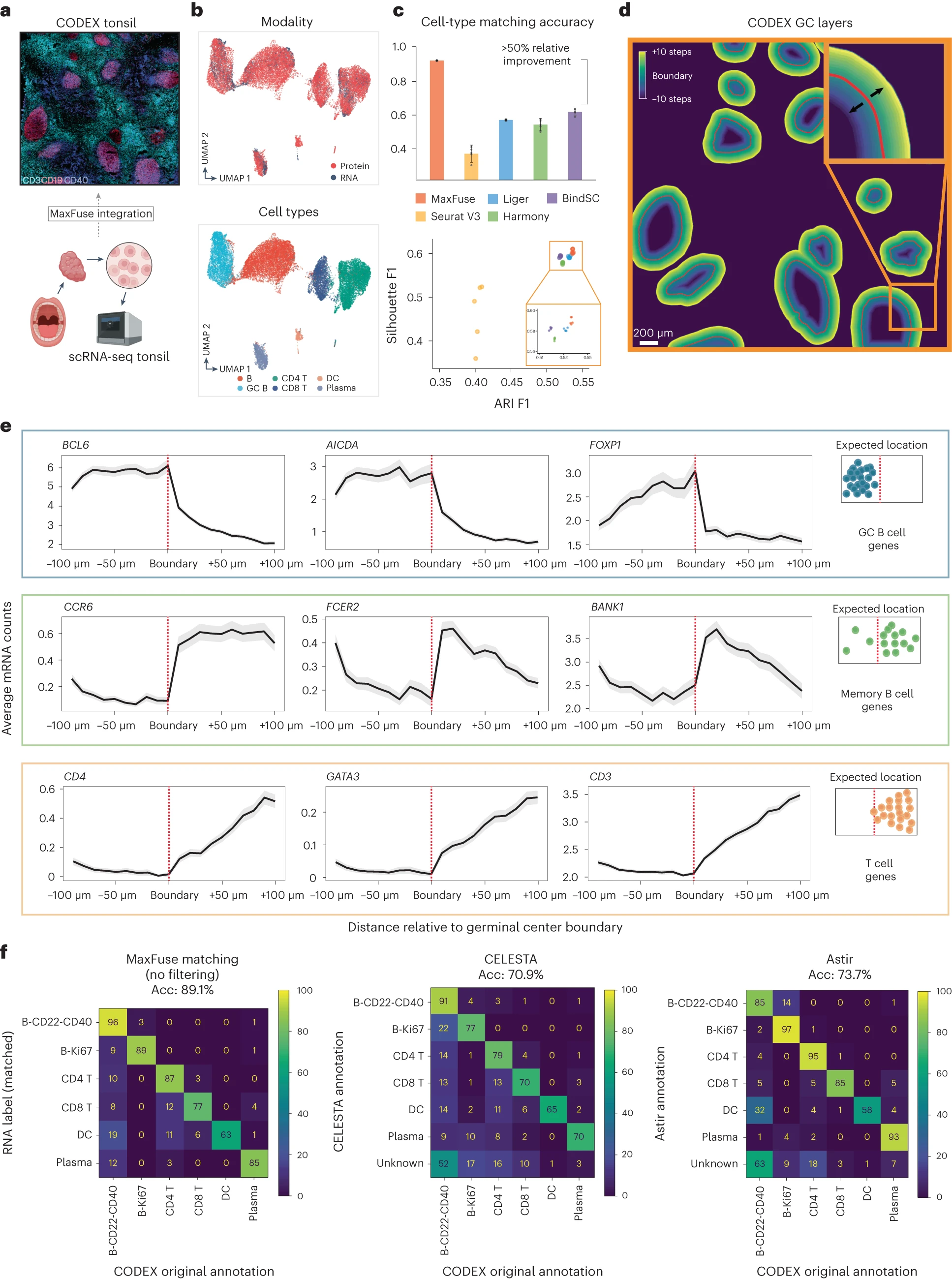
Figure 4. MaxFuse 可发现信息丰富的空间模式
(a) 来自 Kennedy-Darling 等 的 CODEX 数据整合示意图(上图),scRNA-seq 数据来自 King 等 从人类获得(下图)。
(b) 扁桃体 CODEX 和 scRNA-seq 数据的 MaxFuse 集成的 UMAP 可视化,按模态(上图)和细胞类型(下图)着色。
(c) 评估 MaxFuse 和其他方法性能的指标(细胞类型匹配准确性、Silhouette F1 分数和 ARI F1 分数)。
(d) 从GC边界向内/向外延伸的细胞层的图示。
(e) 每层细胞间的平均信使 RNA 计数(通过 MaxFuse 连接)与相对于 GC 边界的层位置(边界左侧向内,右侧向外)进行绘制。
(f) MaxFuse 和其他人类扁桃体 CODEX 数据细胞类型注释方法的基准测试。
# Figure 5: MaxFuse enables tri-modal integration with HUBMAP data.
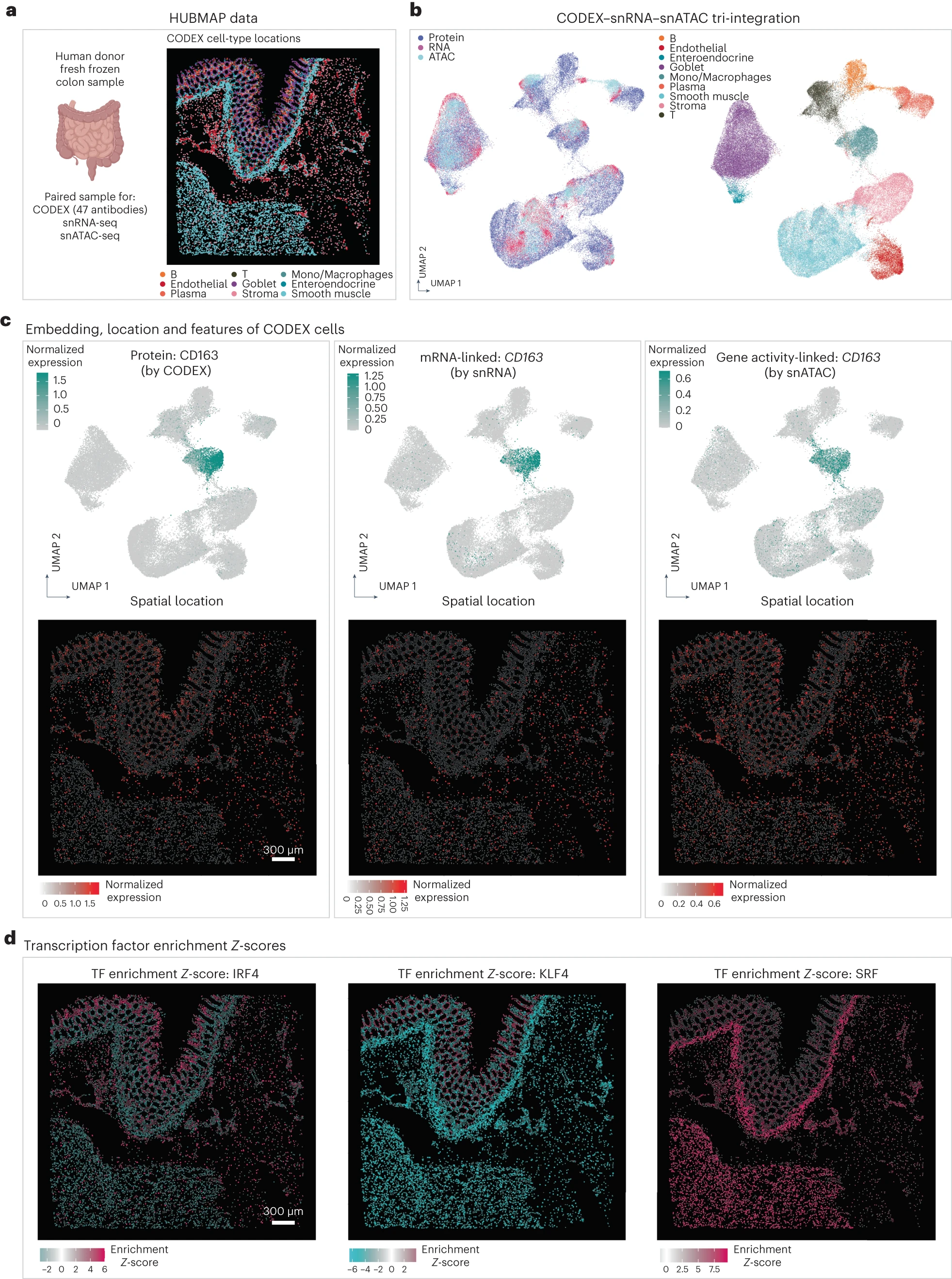
Figure 5. MaxFuse 支持与 HUBMAP 数据的三模态集成
(a) 来自 HUBMAP 联盟的 CODEX、snRNA-seq 和 snATAC-seq 单细胞人类肠道数据概述(左)。基于 CODEX 数据的代表性细胞类型位置(右)。
(b) MaxFuse 生成的三模态整合嵌入的 UMAP 可视化,按模态着色:蛋白质、RNA 和 ATAC(左图),并按细胞类型着色(右图)。
(c) 上排,基于整合嵌入的 CODEX 细胞的 UMAP 可视化,覆盖 CD163 蛋白表达(来自 CODEX 细胞本身,左图),CD163mRNA 表达(来自匹配的 snRNA-seq 细胞,中图)和CD163基因活性评分(来自匹配的 snATAC-seq 细胞,右图)。下排,基于质心x – y位置的 CODEX 单元的空间位置,覆盖有与上排相应面板中相同的表达特征。
(d) 基于质心的x – y位置的 CODEX 细胞的空间位置,覆盖基于其匹配的 snATAC-seq 细胞的转录因子基序富集分数。
# Discussion:
大多数现有的跨模态数据集成方法都是为跨强链接模态的集成而开发的,随着跨模态链接强度的减弱,它们的性能会显着下降。MaxFuse 的动机是弱连锁这一具有挑战性的案例,并专注于弱连锁这一具有挑战性的案例,随着许多新兴研究设计包括带有目标标记面板的空间数据,与单细胞测序数据联合收集,这种情况已经变得越来越普遍。
MaxFuse 依靠两个关键流程来克服弱链接。第一个是“模糊平滑”过程,通过将链接特征的值移向图形平滑值来对它们进行去噪,其中图形由所有特征确定。第二个是迭代细化过程,通过共嵌入、图形平滑和匹配的迭代循环来改进跨模式匹配。这确保了两种模式中所有特征的信息都用于生成最终匹配。我们证明 MaxFuse 极大地改进了最先进的方法,实现了目标蛋白测定数据与转录组和表观基因组水平测定数据的准确整合。MaxFuse的适用性是普遍的。对于强联动场景,MaxFuse 精度与 scGLUE 相当,一种基于深度学习的最先进方法,但计算成本却相当低。此外,当其他集成方法的联合嵌入坐标可用时,这些坐标可以用作 MaxFuse 中的链接特征。轻计算架构以及结合领域知识和现有集成结果的灵活性使得MaxFuse框架适用于广泛的跨模式集成任务。