
 Spatial epigenome-transcriptome co-profiling of mammalian tissues
Spatial epigenome-transcriptome co-profiling of mammalian tissues
题目: 哺乳动物组织的空间表观基因组-转录组联合分析
DOI: https://doi.org/10.1038/s41586-023-05795-1 (opens new window)
Cite: Zhang, D., Deng, Y., Kukanja, P. et al. Spatial epigenome–transcriptome co-profiling of mammalian tissues. Nature 616, 113–122 (2023).
作者介绍:
| Rong Fan |
|---|
 |
| Department of Biomedical Engineering, Yale University, New Haven, CT, USA |
| rong.fan@yale.edu |
# Abstract:
新兴的空间技术,包括空间转录组学和空间表观基因组学,正在成为分析组织环境中细胞状态的强大工具。然而,当前的方法一次只能捕获一层组学信息,排除了检查分子生物学中心法则的机械关系的可能性。在这里,我们提出了两种技术,通过染色质可及性和基因表达,或组蛋白修饰(H3K27me3, H3K27ac或H3K4me3)和基因表达在接近单细胞分辨率的同一组织切片上进行全基因组,表观基因组和转录组的联合分析。这些被应用于胚胎和幼年小鼠大脑以及成人大脑,以绘制表观遗传机制如何控制组织中转录表型和细胞动力学的图谱。尽管通过空间表观基因组或空间转录组鉴定了高度一致的组织特征,但我们也观察到不同的模式,表明它们在定义细胞状态中的不同作用。将表观基因组与转录组逐个像素连接起来,可以揭示组织结构内空间表观遗传启动、分化和基因调控的新见解。这些技术在生命科学和生物医学研究中引起了极大的兴趣。
# Main:
单细胞多组学可以揭示不同组学层的基因调控机制,但缺乏空间信息,这对于理解组织中的细胞功能至关重要。最近出现了空间表观基因组学、转录组学和蛋白质组学但其中大多数只能捕获一层组学信息。尽管计算方法可以整合来自多个组学的数据,但它们不能轻易揭示不同组学层之间的机制联系。此前,我们开发了组织中的确定性条形码,用于空间组学测序,以空间分辨地共同测量转录组和一组蛋白质,最近它也在 10X Visium 平台中实现了。在此,为了进一步研究基因表达调控背后的表观遗传机制,我们通过同时分析染色质可及性和信使 RNA 表达,开发了表观基因组和转录组的空间解析、全基因组组合(使用测序对转座酶可访问的染色质和 RNA 进行空间分析 spatial ATAC–RNA-seq),或组蛋白修饰和 mRNA 表达(使用测序(空间 CUT&Tag–RNA-seq)对目标下的切割和标记和 RNA 进行空间分析(空间 CUT&Tag–RNA-seq);应用于 H3K27me3、H3K27ac 或 H3K4me3 组蛋白修饰)通过确定性cobarcoding在细胞水平上进行组织切片,将spatial-ATAC-seq 或spatial-CUT&Tag 的化学与空间转录组学的化学相整合。我们将这些技术应用于胚胎和幼年小鼠大脑以及成人大脑海马体,以剖析表观遗传和转录状态及其在组织中细胞类型动态调节中的作用。这项工作开辟了空间组学的新领域,并可能为生物和生物医学研究提供前所未有的机会。
# Results:
# Figure 1: Design and evaluation of spatial epigenome–transcriptome cosequencing with E13 mouse embryo.
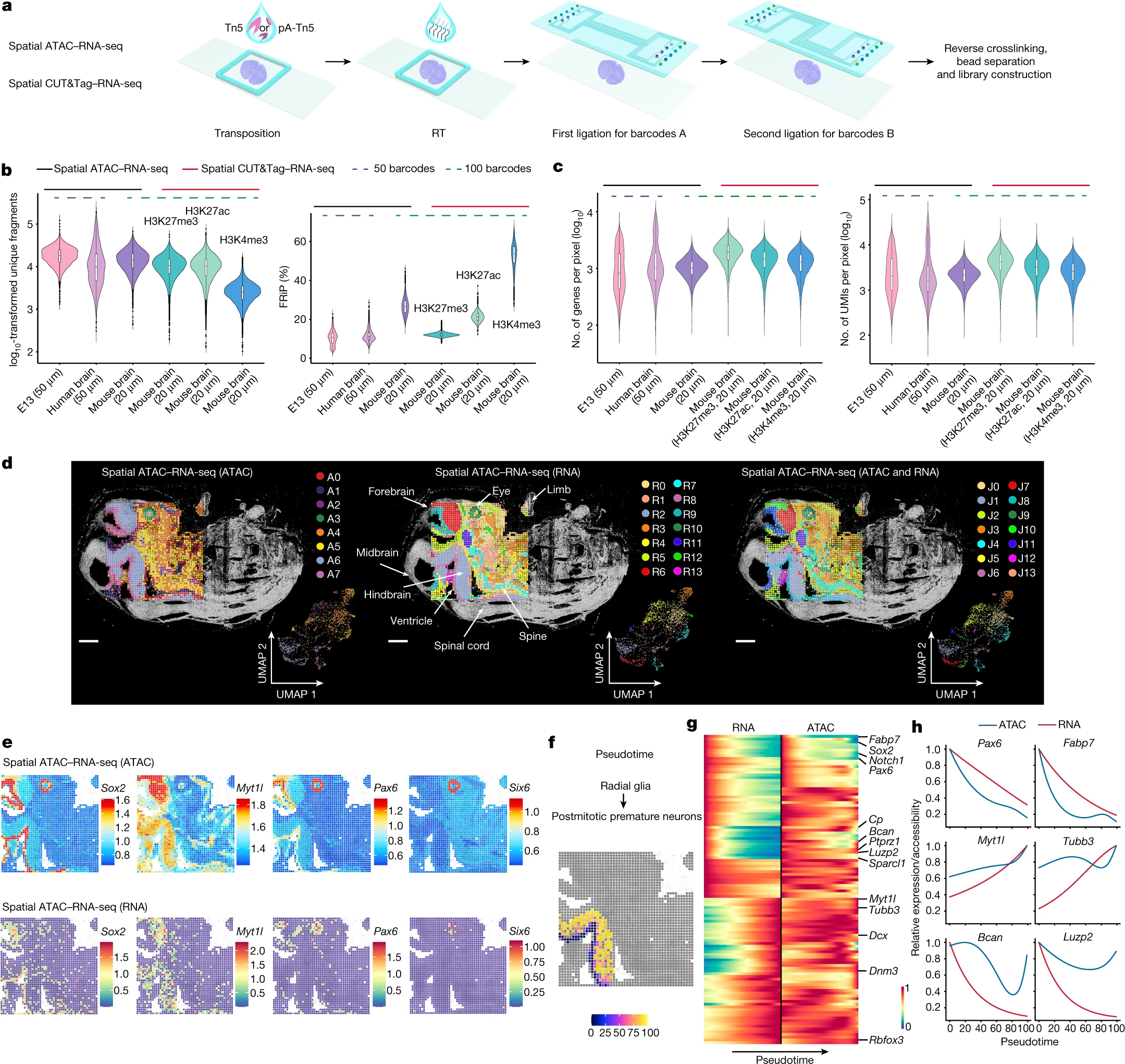
Figure 1. E13小鼠胚胎空间表观基因组-转录组共测序的设计和评估
(a) 工作流程示意图。
(b) 空间 ATAC-RNA-seq 和空间 CUT&Tag-RNA-seq 中独特片段数量和峰读取分数 (FRiP) 的比较。
(c) 空间 ATAC-RNA-seq 和空间 CUT&Tag-RNA-seq 中的基因和 UMI 计数分布。
(d) ATAC、RNA 的所有聚类的空间分布和 UMAP 以及 ATAC 和 RNA 数据的联合聚类。
(e) 空间 ATAC-RNA-seq 中 ATAC 和 RNA 不同簇中所选标记基因的 GAS 和基因表达的空间映射。
(f) 从放射状胶质细胞到有丝分裂后早产神经元的伪时间分析,在空间水平上可视化。
(g) 描绘基因表达和标记基因的 GAS 的热图。
(h) 伪时间内 GAS 和基因表达的动态变化。
# Figure 2: Spatial chromatin accessibility and transcriptome co-profiling of P22 mouse brain.
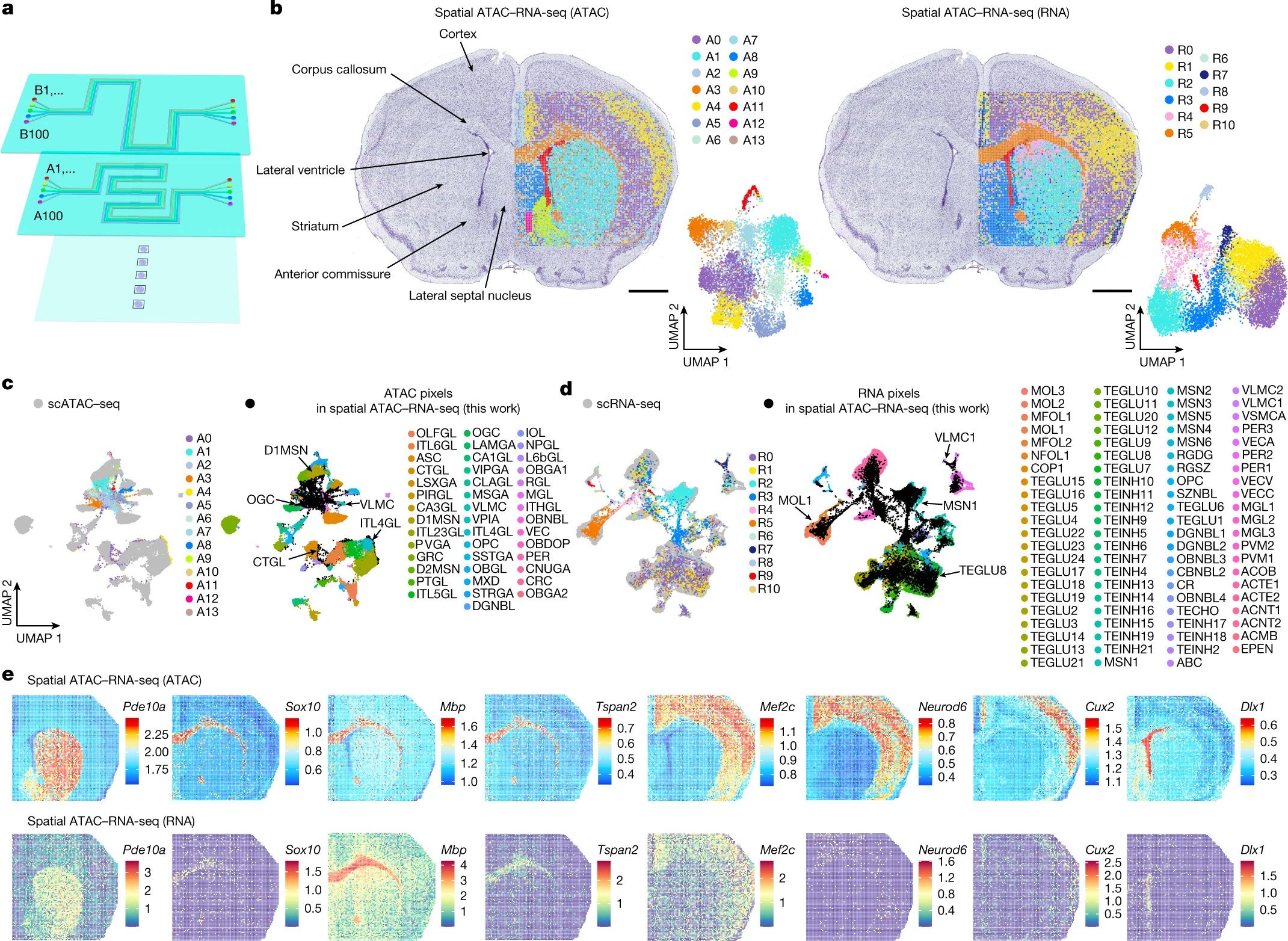
Figure 2. P22 小鼠大脑的空间染色质可及性和转录组联合分析
(a) 通道尺寸为20μm的100×100条码微流控芯片的设计。
(b) 小鼠大脑空间ATAC-RNA-seq中ATAC和RNA所有簇的空间分布和UMAP。
(c) 来自小鼠大脑的ATAC数据和scATAC-seq数据的整合。
(d) 来自小鼠大脑的RNA数据和scRNA-seq数据的整合。
(e) 空间 ATAC-RNA-seq 中 ATAC 和 RNA 不同簇中所选标记基因的 GAS 和基因表达的空间映射。
# Figure 3: Spatial histone modification and transcriptome co-profiling of P22 mouse brain.
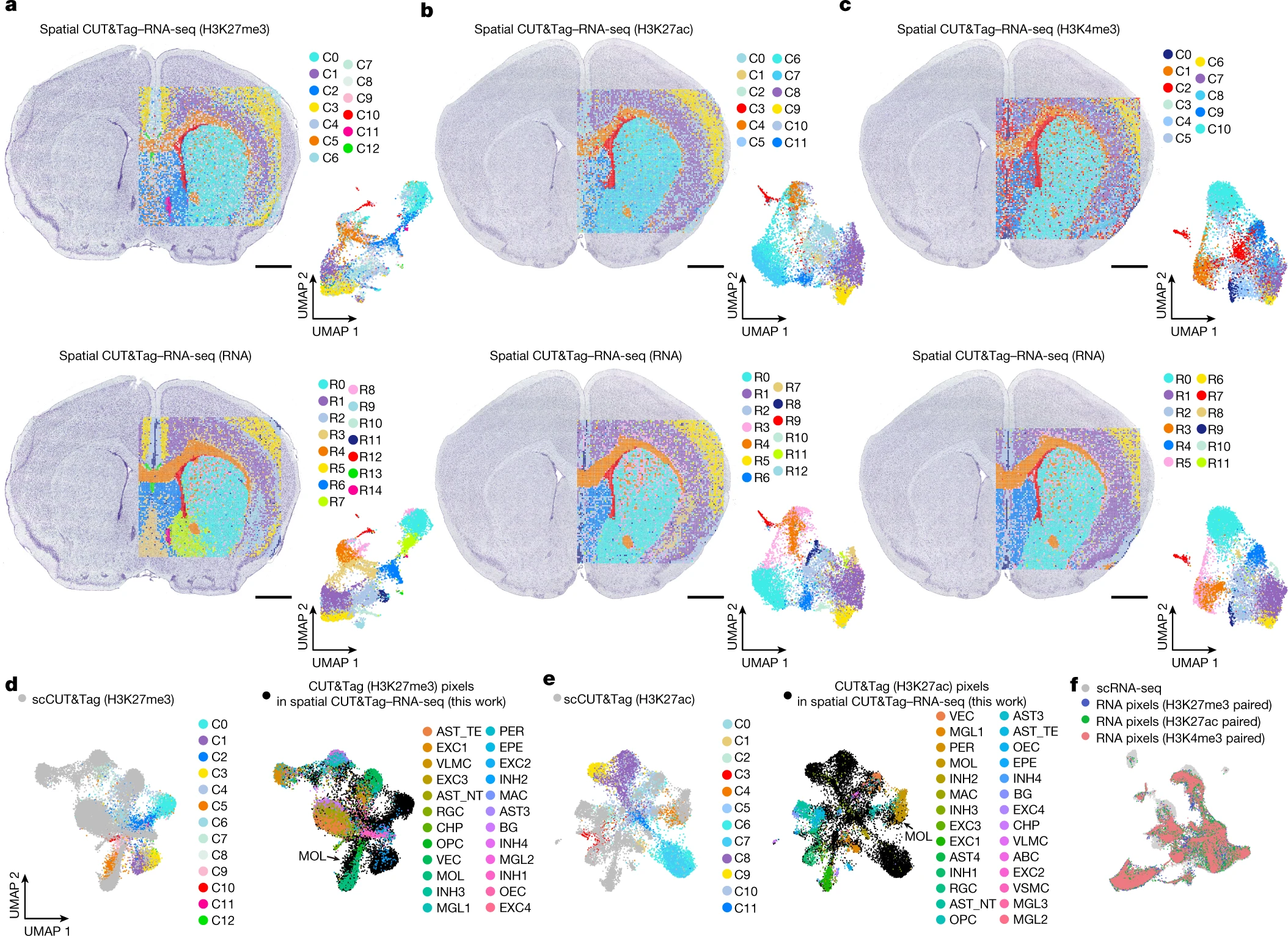
Figure 3. P22 小鼠大脑的空间组蛋白修饰和转录组联合分析
(a-c) 小鼠大脑中H3K27me3 和 RNA ( a )、H3K27ac 和 RNA ( b ) 以及 H3K4me3 和 RNA ( c )所有簇的空间分布和 UMAP 。
(d) H3K27me3 数据与来自小鼠大脑的scCUT&Tag (H3K27me3) 数据的整合。
(e) H3K27ac 数据与来自小鼠大脑的scCUT&Tag (H3K27ac) 数据的整合。
(f) 空间 CUT&Tag (H3K27me3)–RNA-seq、空间 CUT&Tag (H3K27ac)–RNA-seq 和空间 CUT&Tag (H3K4me3)–RNA-seq 中的 RNA 数据与来自小鼠大脑的scRNA-seq 数据的整合。
# Figure 4: Region-specific epigenetic regulation of gene expression.
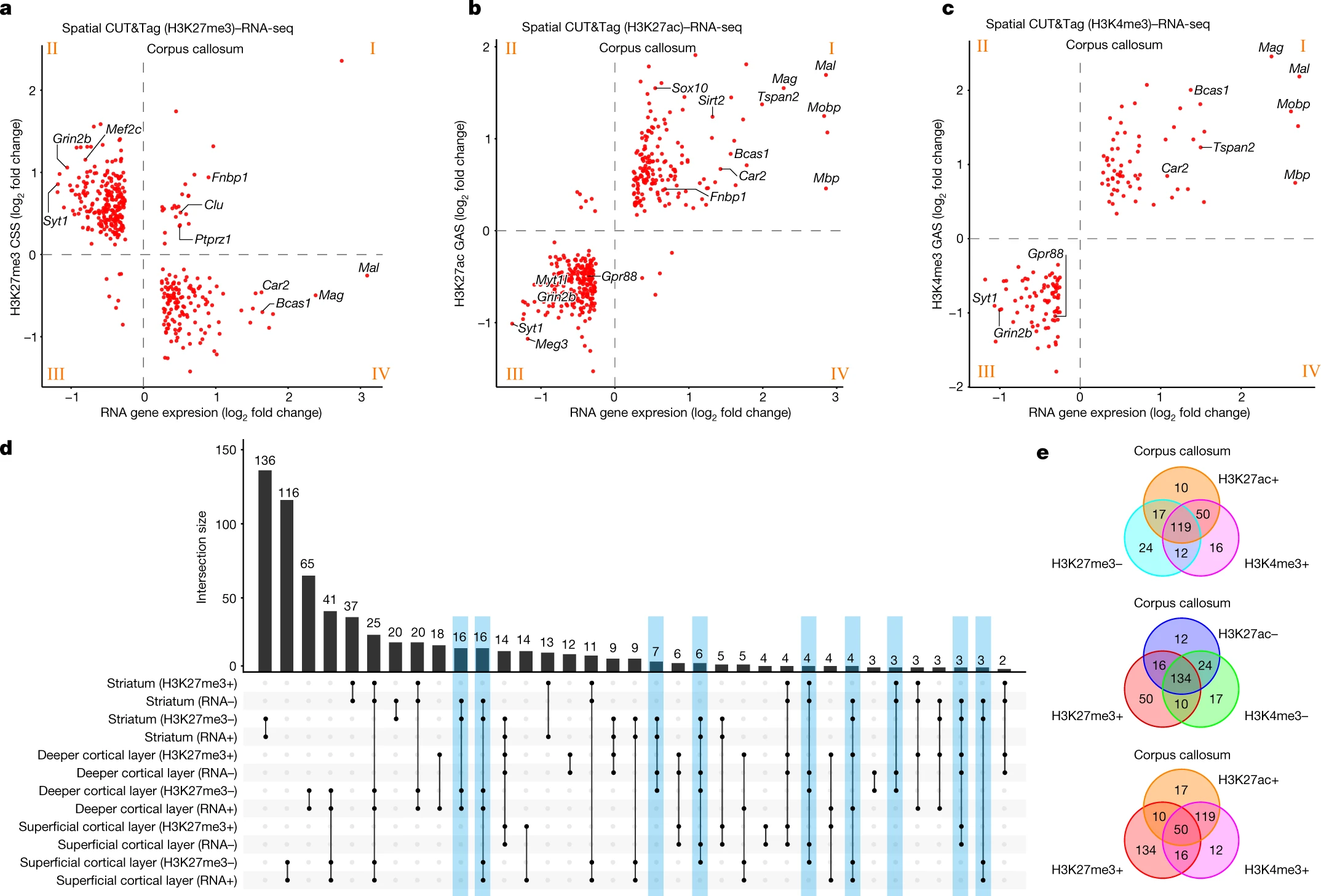
Figure 4. 基因表达的区域特异性表观遗传调控
(a-c) 胼胝体中H3K27me3 CSS 和 RNA 基因表达 ( a )、H3K27ac GAS 和 RNA 基因表达 ( b ) 以及 H3K4me3 GAS 和 RNA 基因表达 ( c ) 的相关性。
(d) 纹状体以及更深和浅层皮质层中 H3K27me3 CSS 和 RNA 基因表达的翻转图。
(e) 维恩图显示具有常见 RNA 标记基因的胼胝体中不同组蛋白修饰的高 (+) 或低 (-) CSS/GAS。
# Figure 5: Spatial chromatin accessibility and transcriptome co-profiling of human hippocampus.
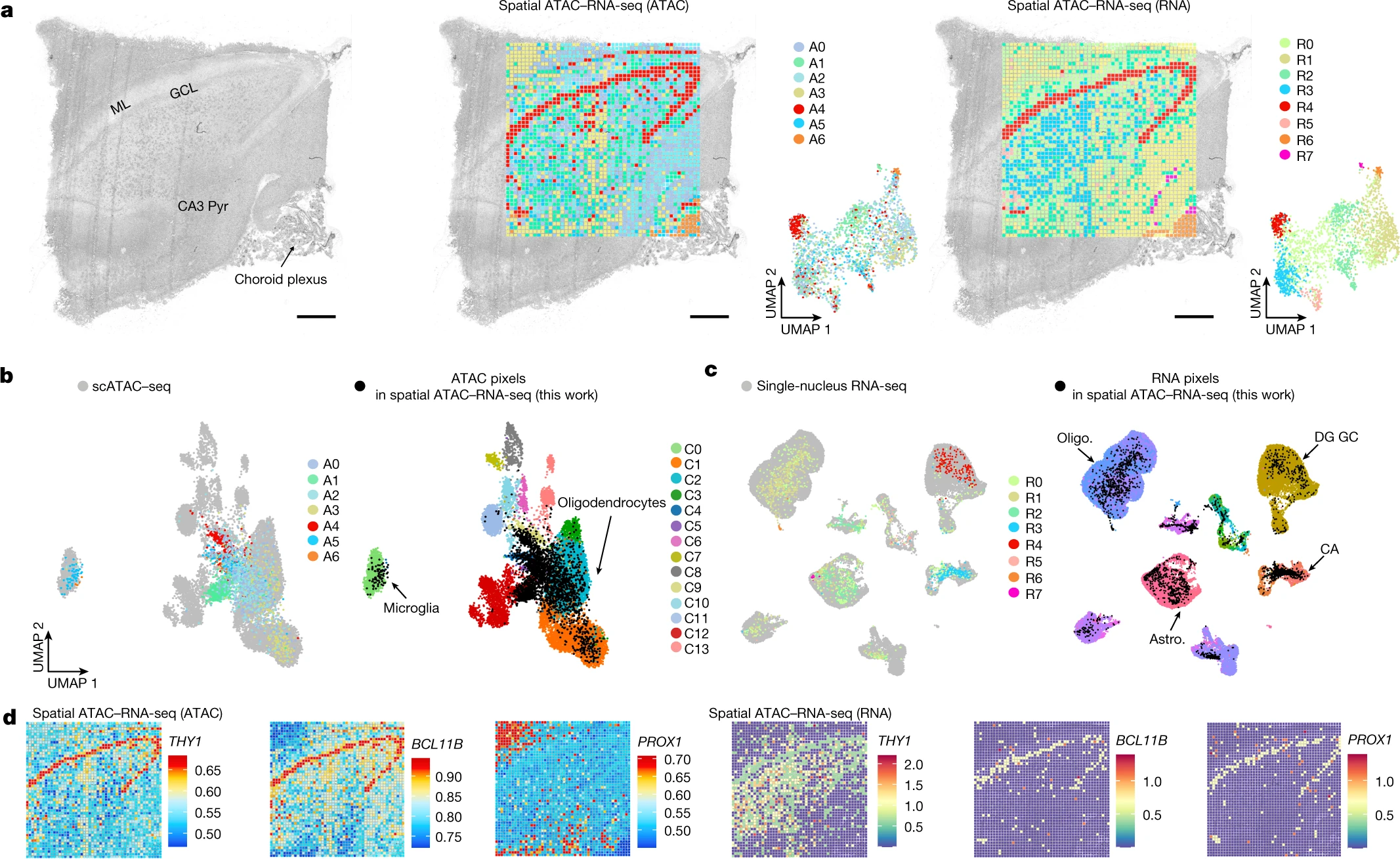
Figure 5.人类海马体的空间染色质可及性和转录组联合分析
(a) 基于人类海马中 ATAC 和 RNA 的所有簇的明场图像、空间分布和 UMAP。
(b) 我们的 ATAC 数据与来自人类海马体的 scATAC-seq 数据的整合。
(c) 将我们的 RNA 数据与来自人脑的 snRNA-seq 数据整合。
(d) ATAC和RNA不同簇中所选标记基因的GAS和基因表达的空间图谱。
# Discussion:
到目前为止,目前的技术还不能在细胞水平上对同一组织切片的表观基因组和转录组进行无偏倚的全基因组比较。我们开发了空间ATAC-RNA-seq和空间CUT&Tag-RNA-seq(适用于H3K27me3, H3K27ac和H3K4me3),用于在接近单细胞分辨率的相同组织切片上与全基因组转录组结合的全基因组染色质可及性或组蛋白修饰共同分析。
总之,在细胞水平上对表观基因组和转录组进行空间分辨、全基因组共测序是空间生物学中信息最丰富的工具之一,可应用于广泛的生物学和生物医学研究。