
 Significance analysis for clustering with single-cell RNA-sequencing data
Significance analysis for clustering with single-cell RNA-sequencing data
题目: 单细胞RNA测序数据聚类的显著性分析
DOI: https://doi.org/10.1038/s41592-023-01933-9 (opens new window)
Cite:Grabski, I.N., Street, K. & Irizarry, R.A. Significance analysis for clustering with single-cell RNA-sequencing data. Nat Methods 20, 1196–1202 (2023).
作者介绍:
| Isabella N. Grabski |
|---|
 |
| Department of Biostatistics, Harvard T. H. Chan School of Public Health, Boston, MA, USA |
| isabellagrabski@g.harvard.edu |
# Abstract:
单细胞 RNA 测序数据的无监督聚类能够识别不同的细胞群。然而,最广泛使用的聚类算法是启发式的,并没有正式考虑统计不确定性。我们发现,不以统计上严格的方式解决已知的变异来源可能会导致对新细胞类型的发现过度自信。在这里,我们扩展了以前的方法,即层次聚类的重要性,提出了一种基于模型的假设检验方法,该方法将显著性分析纳入聚类算法,并允许将簇作为不同的细胞群体进行统计评估。我们还采用这种方法来允许对任何算法报告的集群进行统计评估。最后,我们扩展这些方法来考虑批处理结构。我们针对流行的集群工作流程对我们的方法进行了基准测试,证明了性能的提高。为了展示实用性,我们将我们的方法应用于人类肺细胞图谱和小鼠小脑皮层图谱,识别了几个过度聚类的案例并概括了经过实验验证的细胞类型定义。
# Results:
# Figure 1: Clustering results for applying Seurat’s implementation of the Louvain algorithm to simulated data representing one cell population.
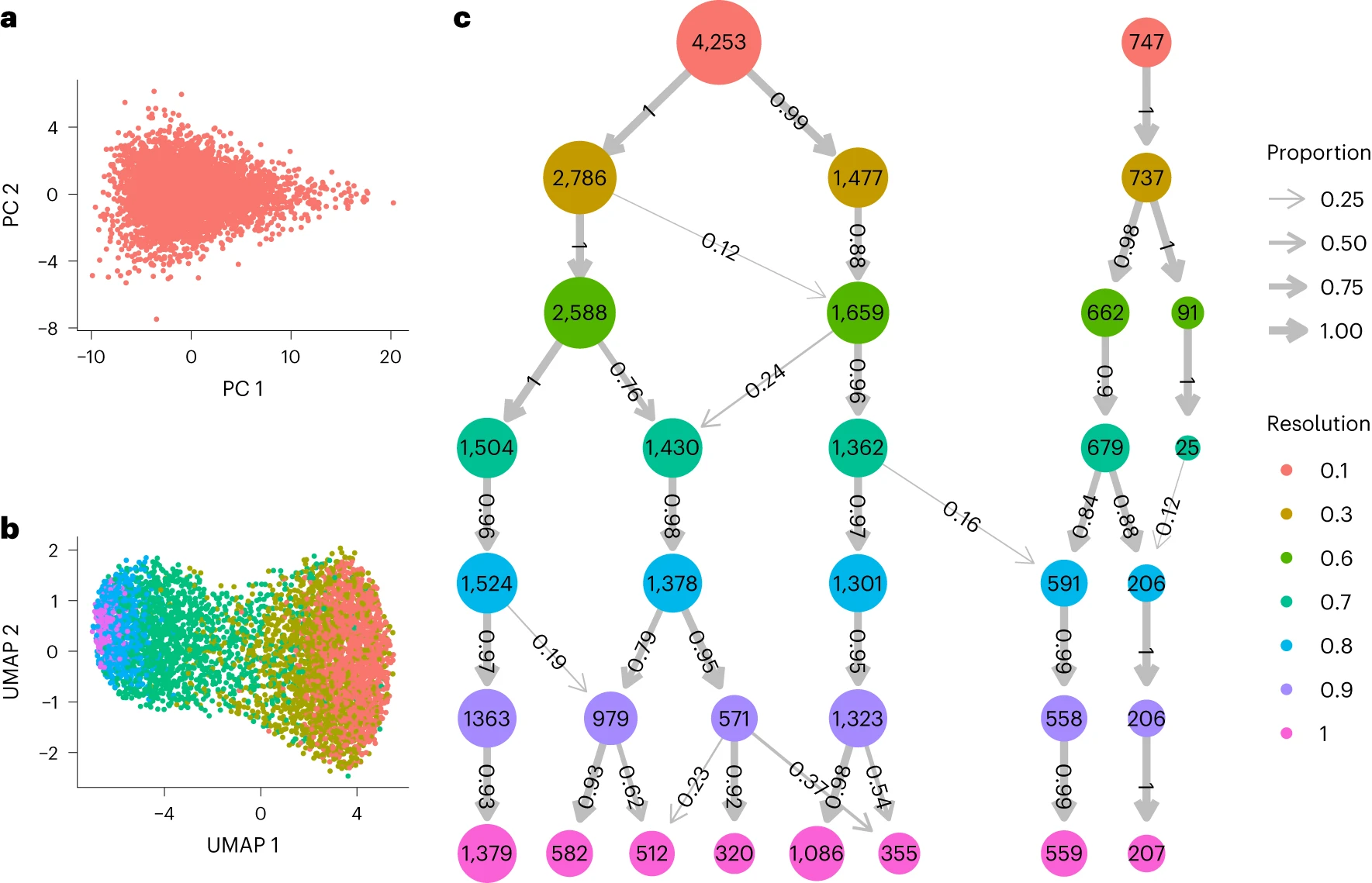
Figure 1. 将 Seurat 的 Louvain 算法实现应用于代表一个细胞群的模拟数据的聚类结果
(a) 由超过 1,000 个基因的相同联合分布生成的 5,000 个细胞的 PCA 图。
(b) 使用默认分辨率参数 0.8 时,UMAP 图按簇成员资格着色。
(c) 使用 Seurat 在不同分辨率参数下实现的 Louvain 算法对结果簇进行修改后的簇树可视化,用行表示。每行中的圆圈数表示使用相应分辨率参数找到的簇数,圆圈的标签和面积表示簇中细胞的数量。箭头宽度和标签表示每个簇中的细胞比例来自先前分辨率参数的簇。
# Figure 2: Schematic illustrating our approach to significance analysis for clustering.
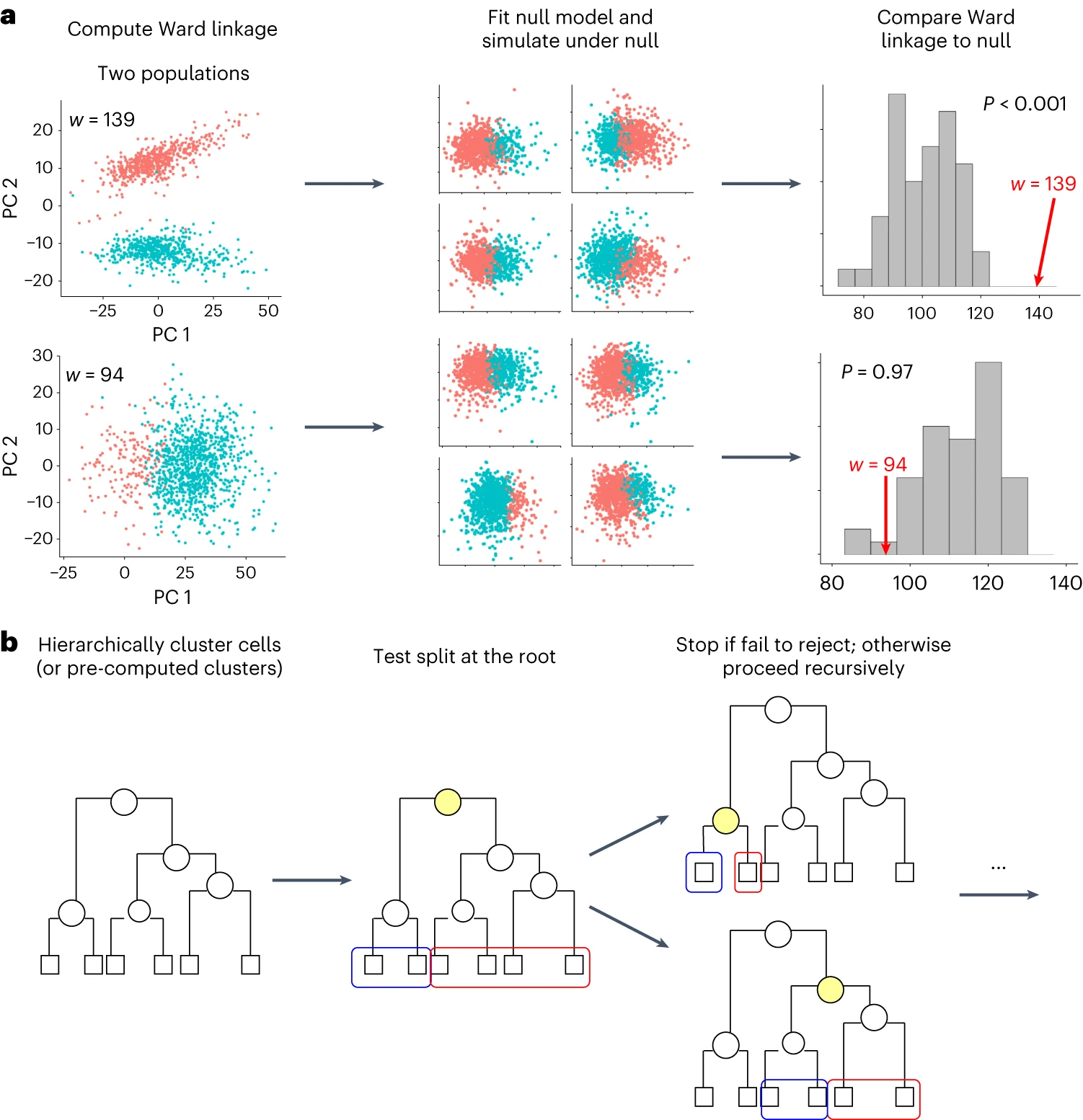
Figure 2. 示意图说明了我们的聚类显著性分析方法。
(a) 我们的方法用于确定所提议的双向拆分是否显着的测试示意图。我们展示了两个示例,其中一个模拟了两个不同的种群(顶部),另一个仅模拟了一个种群(底部)。该示意图显示了我们如何使用层次聚类将数据一分为二,将单个参数模型拟合到数据,在该模型下模拟 100 个数据集,并对每个模拟数据集进行聚类并计算 Ward 关联。然后,我们将观察到的簇的沃德联系与该经验零分布进行比较,以决定是否拒绝原假设。
(b) 我们的方法示意图。我们对所有单元(或预先计算的簇)进行分层聚类,并进行显著性分析以决定是否将根节点拆分为蓝色和红色表示的两个簇。如果我们无法拒绝原假设,我们就会停止,否则递归地继续执行测试来决定是否分割每个节点。
# Figure 3: Additional benchmarks of our approach.
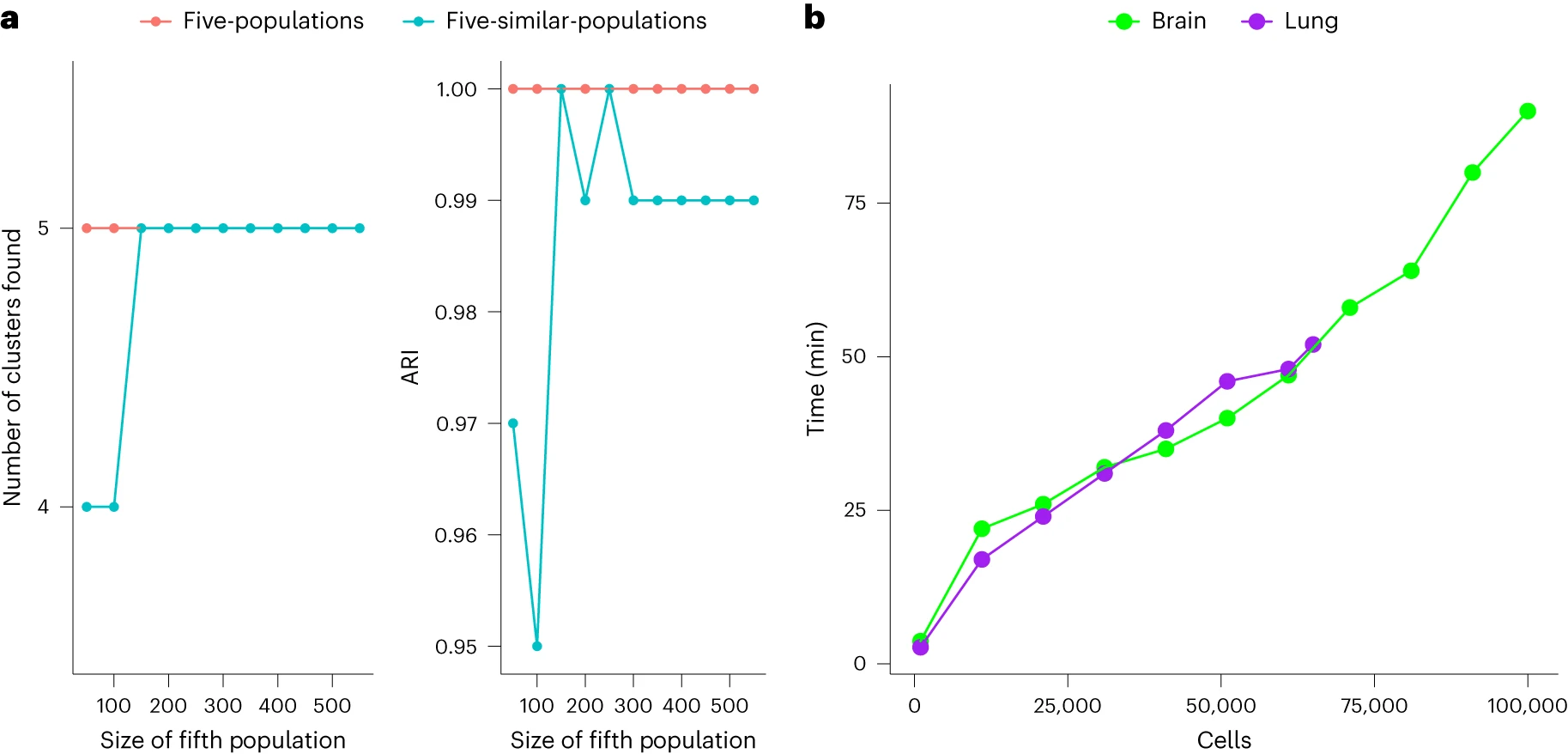
Figure 3. 我们方法的其他基准
(a) 当五个群体和五个相似群体数据集中第五个群体的大小从 50 到 550 个细胞变化时,根据我们的方法发现的簇的数量和 ARI。
(b) sc-SHC 对小鼠小脑数据集(大脑)从 1,000 到 100,000 个细胞的子集的计算时间,以及我们对人类肺细胞图谱(肺)子集的预计算簇的显著性分析的计算时间,范围从 1,000 到 65,000 个细胞。
# Figure 4: Applying significance analysis to clusters reported by the Human Lung Cell Atlas.
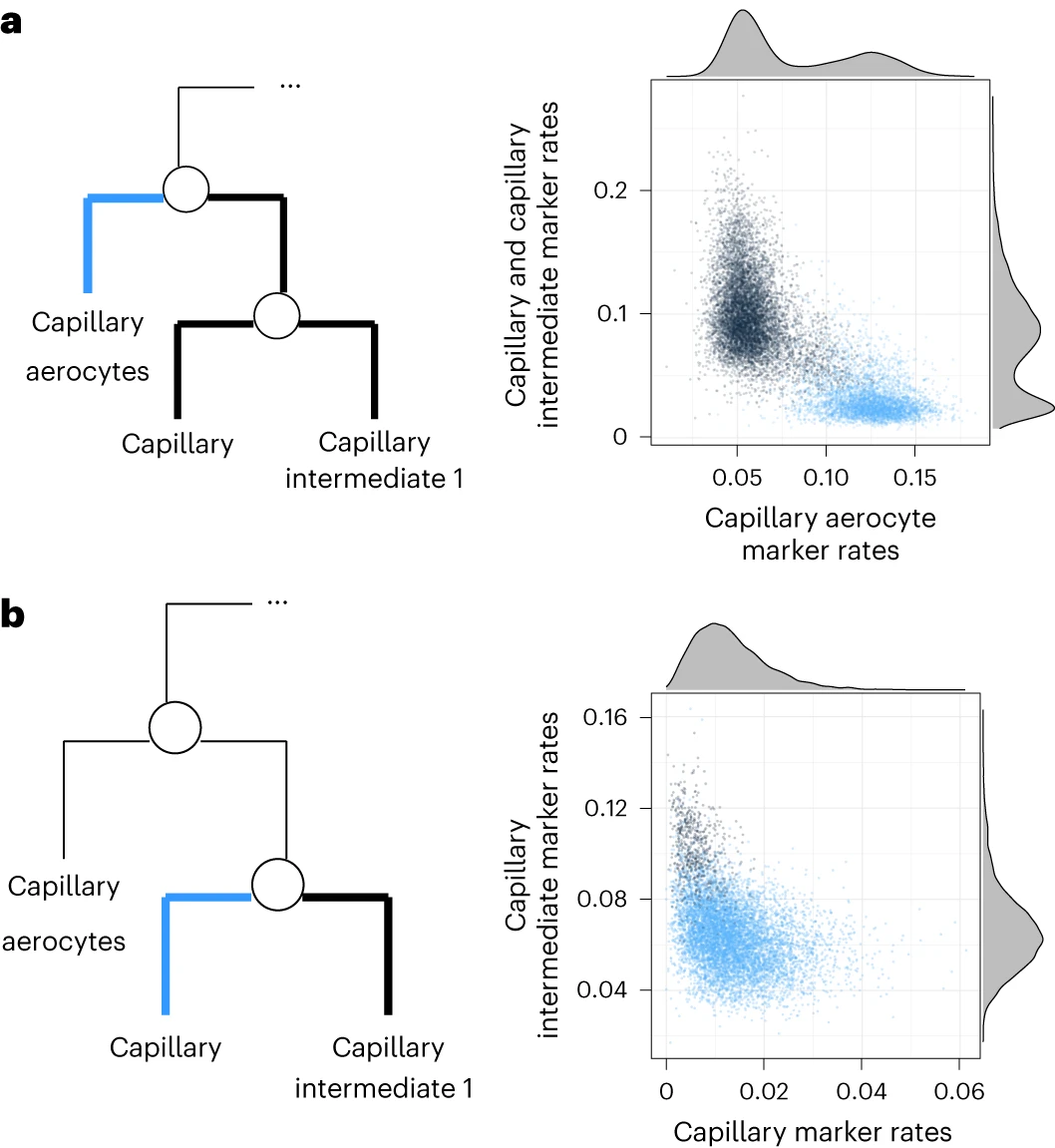
Figure 4. 对人类肺细胞图谱报告的簇应用显著性分析
(a) scRNA-seq 门控图,显示毛细管需氧细胞、毛细管和毛细管中间 1 细胞之间的分裂。我们将每个细胞中代表毛细管和毛细管中间 1 细胞中显着更高表达基因的 UMI 比例与代表毛细管有氧细胞中显着更高表达基因的 UMI 比例进行比较。
(b) 毛细管和毛细管中间 1 细胞之间建议的分裂的 scRNA-seq 门控图。
# Discussion:
聚类分析是众多 scRNA-seq 分析流程中不可或缺的一部分。scRNA-seq 数据受到自然和技术随机变异的影响,但最流行的流程并未考虑统计不确定性。因此,过度聚类很常见,过度自信的解释可能会导致有缺陷的细胞类型注释和错误地声称发现了新的亚型。为了解决这个问题,我们提出了一种将算法聚类与概率模型相结合的显著性分析框架。通过假设基因表达的基本参数模型,我们在先前开发的统计方法的基础上创建了一个参数引导程序,该程序评估即使仅存在一个群体,是否也会出现观察到的聚类结构。尤其,我们提出了应用这个想法的两种方法:一种独立的方法(sc-SHC),它将假设检验构建到层次聚类中,以自动识别与不同细胞群相对应的簇;以及一种事后方法,可以评估任何提供的簇集可能出现过度聚类。我们进一步扩展了这些方法,以适应具有批量效应的更复杂的数据集,例如由于多样本结构。
基于模型的方法的局限性在于,不同群体的定义取决于假设描述基因表达的参数模型的适当性。尽管广泛的数据探索表明我们使用的模型实际上描述了来自不同人群的观察数据,但应考虑该模型不合适的场景。例如,我们假设群体内基因表达呈单峰边缘分布。这意味着,对于某些基因表达遵循多模态分布的细胞群体,我们的方法也可能导致过度聚类,因为不正确的假设可能导致错误地拒绝单一群体零假设。另请注意,与所有聚类算法一样,我们的方法识别离散的细胞群。我们基于模型的方法将这些离散群体与不同的单峰概率分布相关联。然而,这并不排除这些群体中存在具有生物学意义的变异。例如,连续变化的基因表达水平可能与细胞周期内的不同阶段相关。然而,将细胞划分为不同组的方法(例如聚类)并不是统计描述这些重要变异性来源的合适工具,尽管基于模型的方法(例如我们的方法)可以通过模型中的连续参数来量化这种变异性。最后,我们注意到,与所有统计方法一样,我们方法中的任何潜在新颖发现在被接受为事实之前仍应仔细考虑和探索。