
 Single-cell analysis of chromatin accessibility in the adult mouse brain
Single-cell analysis of chromatin accessibility in the adult mouse brain
题目: 成年小鼠大脑染色质可及性的单细胞分析
DOI: https://doi.org/10.1038/s41586-023-06824-9 (opens new window)
Cite: Zu, S., Li, Y.E., Wang, K. et al. Single-cell analysis of chromatin accessibility in the adult mouse brain. Nature 624, 378–389 (2023).
作者介绍:
| Bing Ren |
|---|
 |
| Department of Cellular and Molecular Medicine, University of California San Diego, School of Medicine, La Jolla, CA, USA |
| biren@health.ucsd.edu |
# Abstract:
最近单细胞技术的进展使我们发现了成千上万种脑细胞类型;然而,我们对这些细胞类型中基因调控程序的理解远未完善。在这里,我们报告了一份成年小鼠大脑的候选顺式调控DNA元件(cCREs)的全面图谱,通过分析来自117个解剖学切片的230万个单个脑细胞的染色质可及性而生成。该图谱包括约100万个cCRE以及它们在1,482个不同的脑细胞群体中的染色质可及性,为小鼠基因组最新的此类注释增加了超过446,000个cCRE。小鼠大脑的cCRE在人类大脑中有适度的保守性。小鼠特有的cCRE,具体来说,是从一部分皮质兴奋性神经元中鉴定出来的,强烈富集了转座元件,暗示了转座元件在新的调控程序和神经元多样性的出现中可能发挥作用。最后,我们推断了260多个小鼠脑细胞亚类的基因调控网络,并开发了深度学习模型,能够仅从DNA序列中预测不同脑细胞类型中基因调控元素的活动。我们的结果为分析小鼠和人类大脑中细胞特异性基因调控程序提供了资源。
# Main:
顺式调控元件(CREs)通过与序列特异性转录因子(TFs)的结合以及染色质重塑蛋白和转录机制的募集,控制基因在时空上的表达。这些元件,包括启动子、增强子、隔离子、沉默子以及其他较少被了解的调控序列,共同协作推动在发育、分化和疾病中的细胞特异性基因表达。为了更全面地描绘小鼠脑细胞中的cCREs,我们使用了单核酸酶切可及染色质测序(snATAC–seq)技术,在整个成年小鼠大脑中以单细胞分辨率分析染色质可及性。
# Results:
# Figure 1: Single-cell analysis of chromatin accessibility in the adult whole mouse brain.
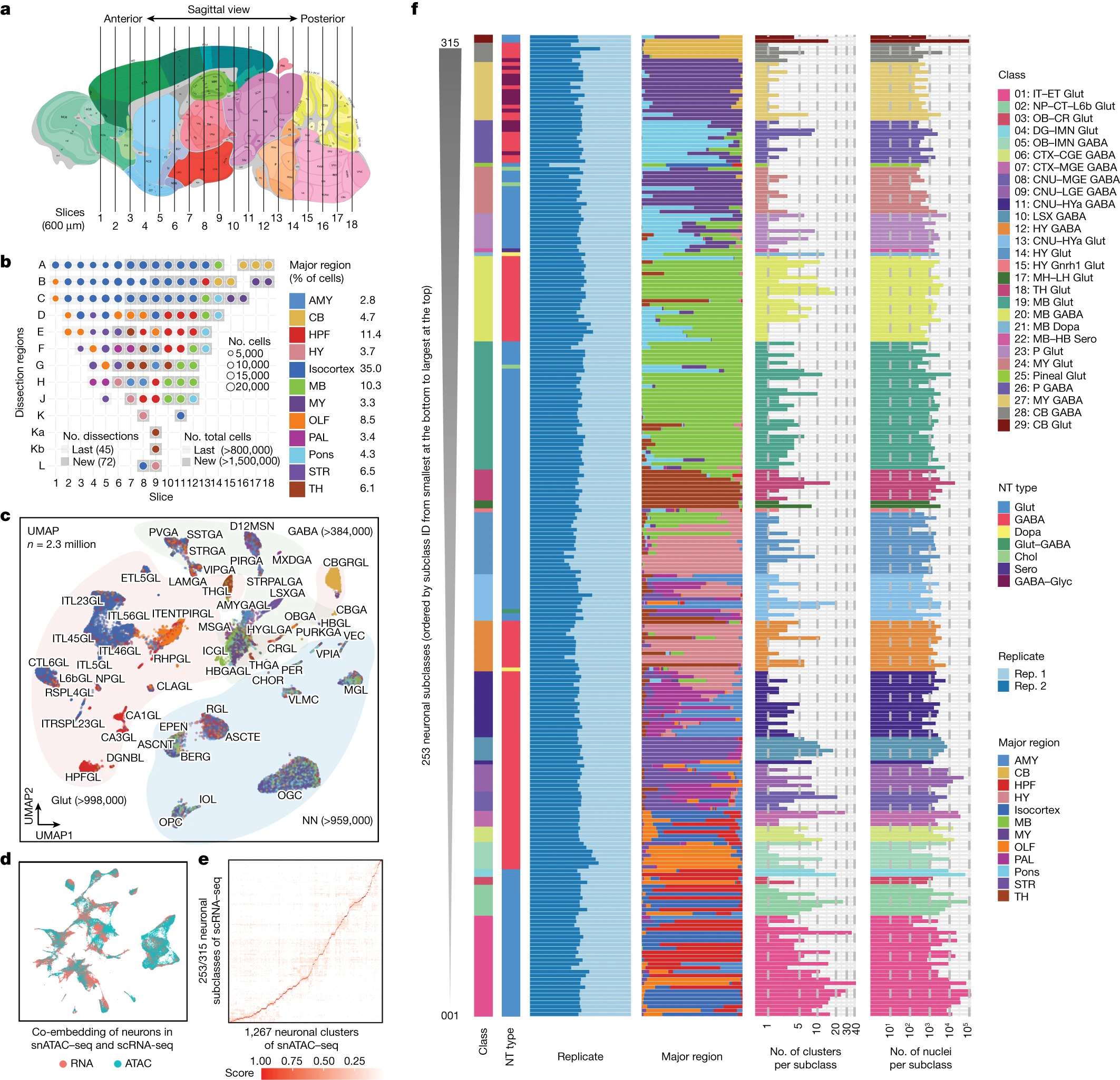
Figure 1. 成年小鼠全脑染色质可及性的单细胞分析
(a) 示例剖析策略示意图。
(b) 质量控制和双细胞去除后 117 次解剖的细胞核数量。
(c) snATAC-seq数据的UMAP嵌入和聚类分析。
(d) scRNA-seq数据和snATAC-seq数据的神经元细胞的共嵌入UMAP嵌入,按两种模态着色。
(e) scRNA-seq数据中的神经元亚类与我们的snATAC-seq数据中的L4级神经元簇之间的共识得分。
(f) 我们的snATAC-seq数据中的253个神经元亚类与上述scRNA-seq中的神经元亚类相匹配,并根据亚类ID排序。从左到右,条形图表示类别、主要神经递质(NT)类型、核的生物复制分布、核的主要区域分布、簇的数量和核的数量。
# Figure 2: Identification and characterization of cCREs across mouse brain cell types.
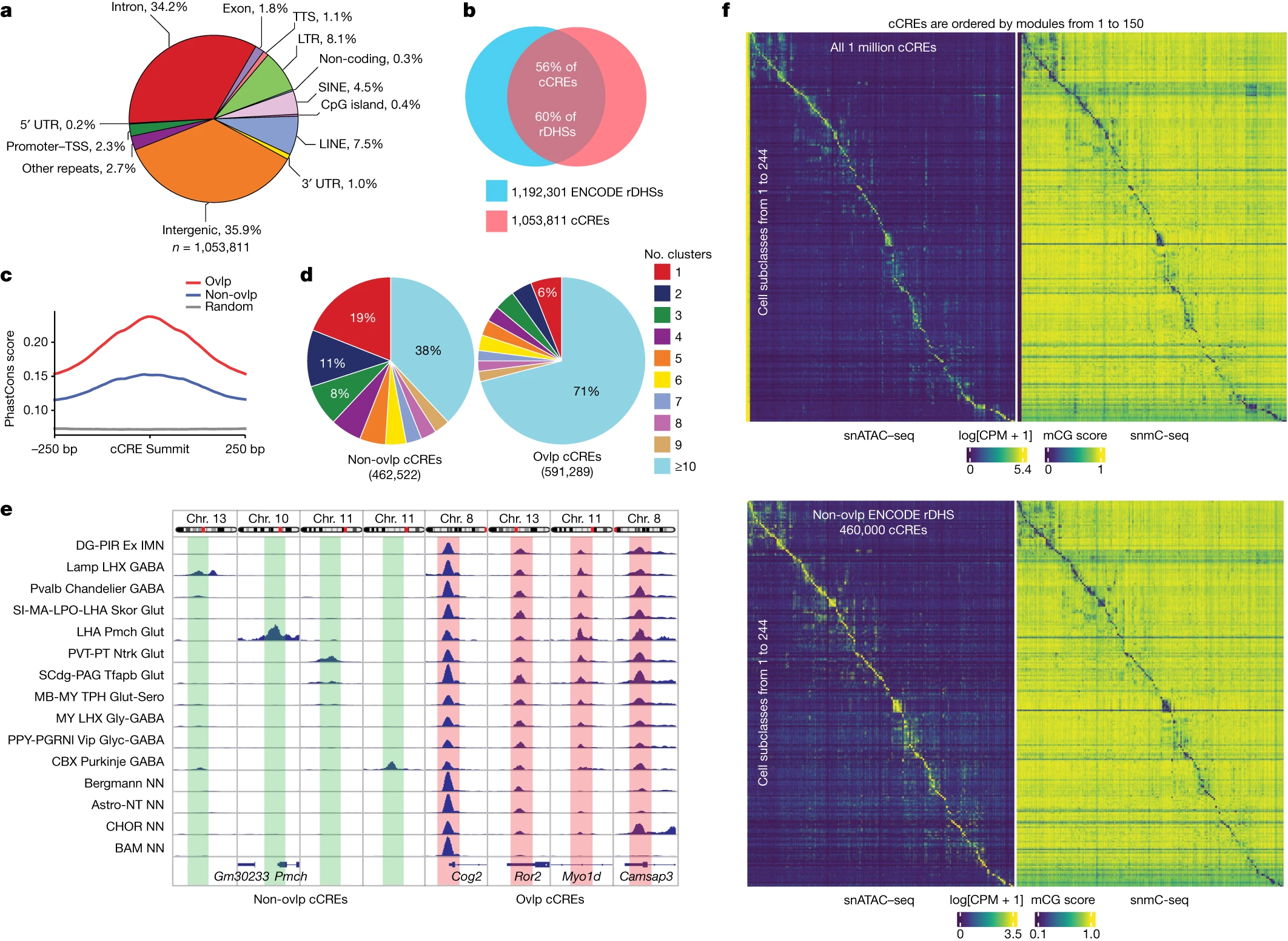
Figure 2. 跨小鼠脑细胞类型的 cCRE 的识别和表征
(a) 使用HOMER确定与小鼠基因组中的注释序列重叠的cCREs的分数。TTS代表转录终止位点,UTR代表非翻译区域。
(b) 本研究中cCREs(红色)与SCREEN数据库中代表性DHSs(蓝色)之间的重叠。
(c) 使用deepTools确定与rDHSs重叠的cCREs(红色),与未与rDHSs重叠的cCREs(蓝色)以及随机基因组背景(灰色)的平均PhastCons保守性分数。
(d) 不同细胞亚型对 peak 值调用的cCREs的比例。左侧,与rDHSs无重叠的cCREs。右侧,与rDHSs有重叠的cCREs。
(e) 两种类型的cCREs的基因组浏览器轨迹。左侧,与rDHSs无重叠的cCREs。右侧,与rDHSs有重叠的cCREs。
(f) 对于snATAC-seq数据中244个共享的细胞亚类的150个顺式调控模块的染色质可及性(左上)。行代表亚类,列为从每个模块中抽样的代表性cCREs。右侧,热图显示来自snmC-seq分析的snDNA甲基化信号,位于相应cCREs的基因组位置,属于相同的亚类。底部,类似于上述的热图,但仅适用于与ENCODE rDHSs无重叠的460,000个cCREs。
# Figure 3: Integrative analysis to identify the potential enhancer–gene connections across the whole mouse brain.
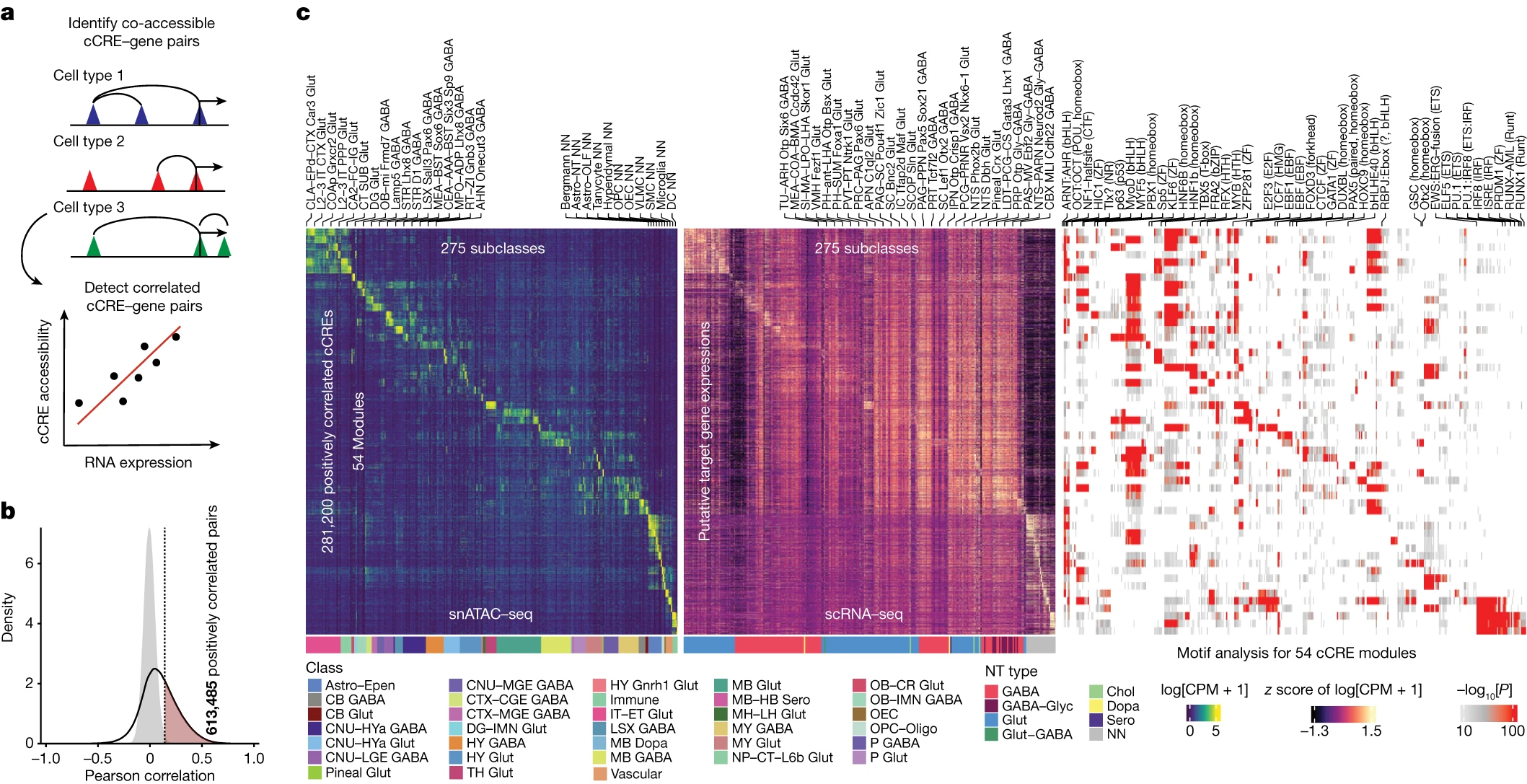
Figure 3. 通过综合分析确定整个小鼠大脑中潜在的增强子-基因连接。
(a) 用于识别与目标基因的mRNA表达呈正相关的cCREs的计算策略示意图;在snATAC–seq和scRNA-seq数据之间,通过对275个细胞亚类计算PCCs。对于每个细胞亚类,使用Cicero预测了共同可及的顺式调控DNA相互作用。
(b) 总共识别了613,485对(红色)呈正相关的cCRE–基因对。灰色填充的曲线显示了对随机洗牌的cCRE–基因对的PCC分布。
(c) 潜在增强子的染色质可及性(左侧);275个细胞亚类中整个小鼠脑中连接基因的mRNA表达(中间);以及不同增强子基因模块中已知TF motif 的富集(右侧)。
# Figure 4: Inference of subclass-specific GRNs across the whole mouse brain.
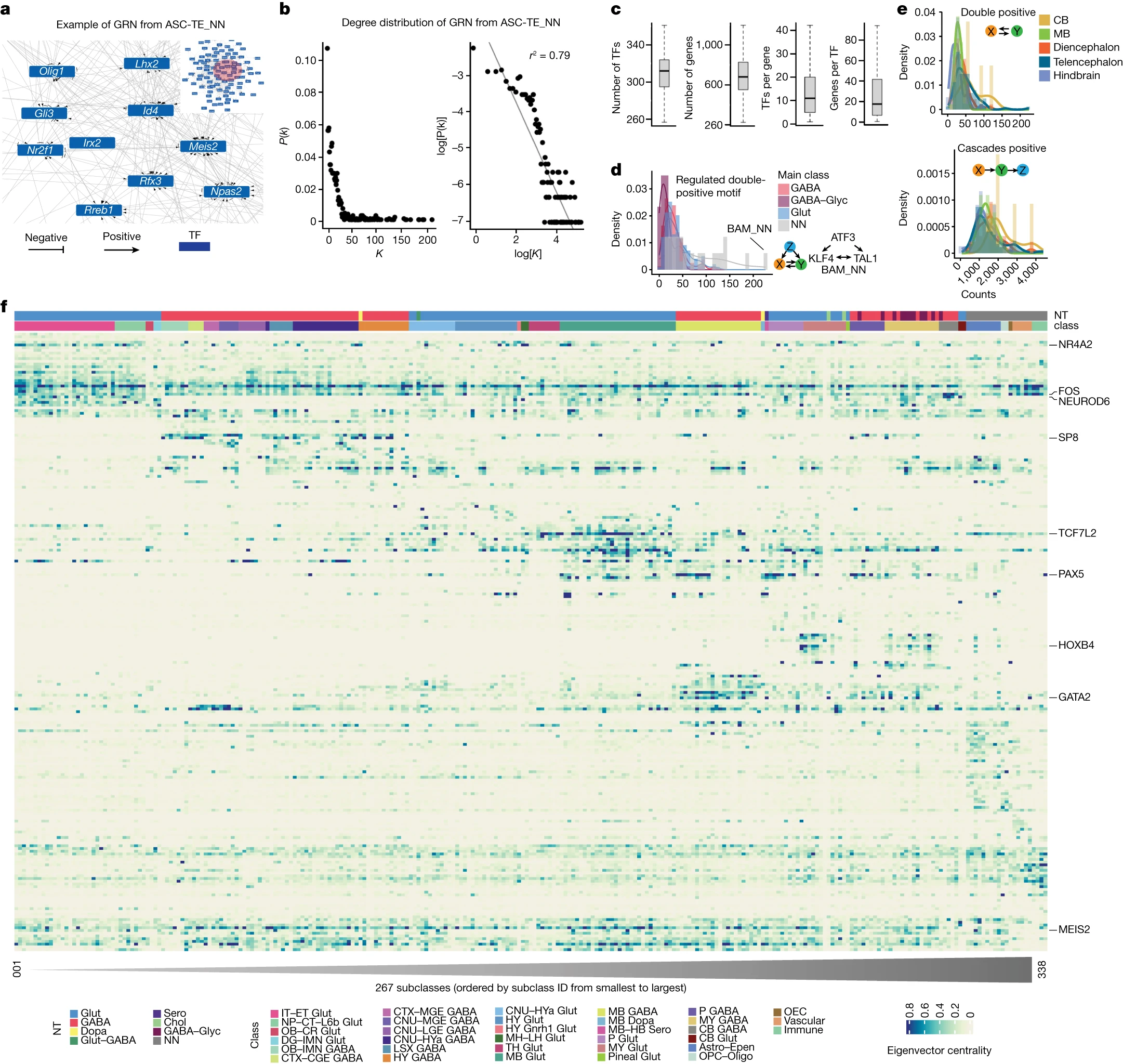
Figure 4. 整个小鼠大脑中亚类特异性 GRN 的推断
(a) 使用CellOracle在大脑皮层区域星形胶质细胞(ASC-TE_NN)中推断的基因调控网络(GRN)示例。边缘带有权重并具有方向,反映了疑似的调控强度和模式(抑制或激活)。
(b) GRN的度分布图。P(k)表示GRN中具有k度的节点的概率。一个节点的度是与它链接的其他节点的数量。
(c) 267个细胞亚类的GRNs中TF的数量,基因的数量,每个基因受调控的TF的数量以及受TF调控的基因的数量。
(d) 每个主要细胞类别的受调控双阳性网络模体的归一化直方图。线是适用于不同直方图的基于核密度的密度曲线。
(e) 五个小鼠脑区域的两个网络模体的直方图:大脑皮层(isocortex、OLF、HPF、STR、PAL和AMY)、间脑(TH和HY)、脑干、后脑(MY和pons)和小脑。
(f) 每个亚类特异性GRN中TF的基于特征向量的中心性或重要性分数的热图。每行代表一个TF,每列代表一个亚类。
# Figure 5: Analyses of chromatin accessibility at TEs of cCREs.
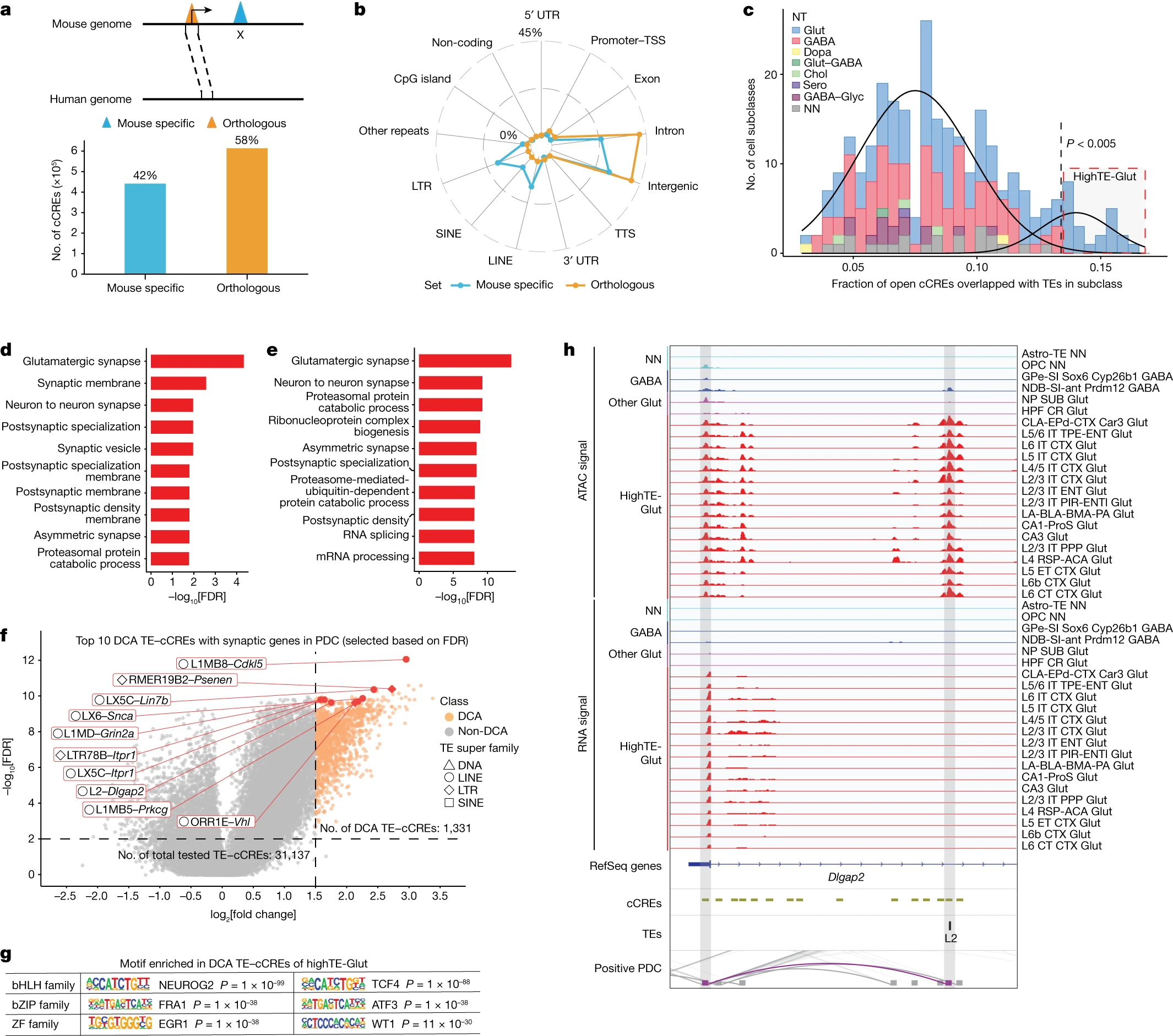
Figure 5.cCRE 的 TE 染色质可及性分析
(a) 鼠标特异性和同源性cCREs的示意图。条形图显示了鼠标特异性和同源性cCREs的数量。
(b) 鼠标特异性和同源性cCREs的基因组分布比例。
(c) 在Glut神经元、GABA能神经元、多巴胺能神经元、乙酰胆碱能神经元、5-羟色胺能神经元、甘氨能神经元和非神经元的每个亚类中,cCREs与转座子的重叠比例。两条曲线显示了混合模型的高斯分布。highTE-Glut指的是其cCREs中高比例与转座子重叠的Glut神经元亚类。
(d) GO分析显示在高TE-Glut亚类中,与TE-cCREs(与转座子相关的cCREs)呈正相关的基因中富集了与神经元特异性功能相关的GO条目,与所有亚类中与TE-cCREs呈正相关的基因相比。
(e) GO分析显示在高TE-Glut亚类中,与TE-cCREs呈正相关的基因中富集了与神经元特异性功能相关的GO条目,与高TE-Glut亚类中所有cCREs呈正相关的基因相比。
(f) 与其他亚类相比,在高TE-Glut亚类中的TE-cCREs上的DCA。显示与突触相关基因相关的前十个DCA TE-cCREs。前十个DCA TE-cCRE–基因对(例如L1MB8–Cdkl5)由红色框表示。前十个DCA TE-cCREs的超级家族由不同形状表示。
(g) 在高TE-Glut神经元中富集的DCA TE-cCREs中的前三个 motif 家族。
(h) NN、GABA、highTE-Glut和其他Glut亚类的选定DCA TE-cCREs和基因对的聚合染色质可及性轨迹的基因组浏览器轨迹。
# Figure 6: Deep-learning models predict chromatin accessibility in different brain cell types from the DNA sequence.
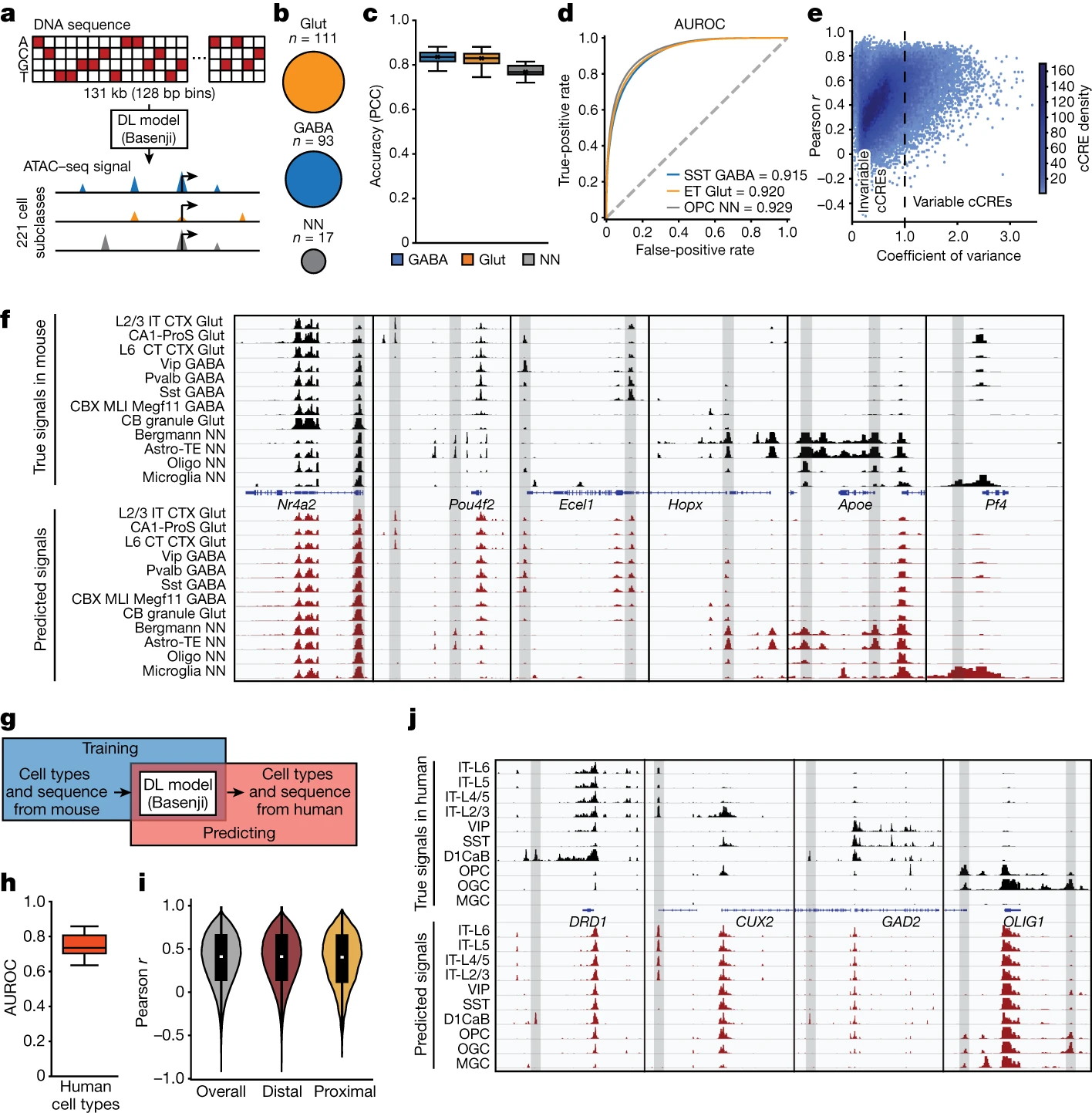
Figure 6.深度学习模型根据 DNA 序列预测不同脑细胞类型的染色质可及性
(a) 用于预测染色质可及性的深度学习(DL)模型Basenji的示意图。
(b) 训练数据集中每个细胞类别的子类数量。
(c) 每个类别的准确性(Pearson相关性)。n = 93(GABA),n = 111(Glut)和n = 17(NN)个子类。
(d) 通过比较从预测的基因组信号调用的 peak 值与从真实实验信号调用的 peak 值,计算了代表性子类的AUROC。
(e) 模型预测细胞类型特异性开放染色质的能力。跨细胞类型的变异系数(方差/均值)与在细胞子类之间计算的真实信号和预测信号之间的Pearson r进行比较。
(f) 在测试集中,通过比较鼠标细胞亚类中的ATAC–seq数据中的真实信号与预测的染色质可及性。显示了Nr4a2、Pou4f2、Ecel1、Hopx、Apoe和Pf4附近的代表性位点。
(g) 使用人类DNA序列作为输入预测潜在染色质可及性信号的示意图。
(h) 通过比较从预测的基因组信号调用的 peak 值与从实验信号调用的 peak 值,计算了匹配的人类细胞类型的AUROC。
(i) 对所有测试的cCREs、测试的远端cCREs和测试的近端cCREs计算的真实信号和预测信号之间的Pearson。
(j) 在人类细胞类型中从ATAC–seq分析中捕获的真实信号和预测的染色质可及性在CUX2、GAD2、DRD1和OLIG1附近的代表性基因组位点上显示。突显了细胞类型特异性的cCREs。
# Discussion:
1.在这里,我们通过对成年小鼠大脑中117个解剖区域的超过230万个细胞进行单细胞染色质可访问性分析,描述了小鼠大脑的全面cCRE目录。
2.我们尝试使用CellOracle对从成年小鼠大脑中收集的单细胞ATAC-seq和RNA-seq数据重建260多个不同大脑细胞亚类的基因调控网络(GRN)。
3.我们通过将本研究中定义的小鼠大脑cCRE图谱与另一项对42个成年男性供体的人脑区域进行的snATAC-seq分析获得的人脑cCRE图谱进行比较,调查了整个小鼠大脑基因调控元件的序列保守性。我们发现,本研究中定义的cCRE约有22%在序列和染色质可及性上在人脑中保守。与先前的报告一致,具有染色质可及性保守性的cCRE倾向于是启动子或远端元件(可能是增强子),并在更广泛的细胞类型中显示可及性。相反,小鼠特异性的cCRE强烈富集于转座子元件,暗示了TE在小鼠大脑中细胞特异性基因表达模式中的潜在作用。我们的结果引发了这样一个有趣的可能性,即在进化过程中,转座子可能对神经回路多样性产生影响。
4.通过从DNA序列中提取上下文信息,深度学习方法最近已被用于预测各种基因组功能特征,如表观遗传修饰、三维相互作用和基因表达。我们采用这种方法开发了基于序列的模型,用于预测275个小鼠大脑细胞亚类的染色质可及性。